4H综合征的临床表现与分子诊断(附1例报告)
2017-11-03梁超陆海英郭虎
梁超,陆海英,郭虎
·学术交流·
4H综合征的临床表现与分子诊断(附1例报告)
梁超,陆海英,郭虎
目的探讨4H综合征的临床表现与分子诊断。方法回顾性分析1例基因诊断明确的4H综合征患者的临床资料,并结合文献资料分析其临床特点。结果患儿,男,6岁8个月,双手抖动1年,自幼精神、运动发育落后,行走不稳,出牙延迟。眼科检查示近视、视神经萎缩。头颅MRI示两侧大脑半球广泛对称性脑白质病变。基因检查示POLR3A复合杂合变异(c.1781T>G,c.2693delT)。诊断为4H综合征。结论4H综合征早期临床表现为精神、运动发育迟缓、出牙延迟,主要临床特点为脑白质髓鞘化不良、近视、共济失调,基因学特点为POLR3A或POLR3B突变。
4H综合征;临床特点;基因学特点
4H综合征又名髓鞘发育不良性脑白质营养不良伴或不伴少牙畸形和/或低促性腺激素性性腺功能减退综合征(hypomyelinating leukodystrophies with or without hypodontia and/or hypogonadotropic hypogonad-ism syndrome)是一种少见的遗传性脑白质病,国内尚未见报道。现就本院收治的1例基因确诊的4H综合征患者的临床资料,结合文献讨论并总结该病的临床与基因学特点,以提高国内对该病的认识。
1 临床资料
患儿,男,6岁8个月,因“双手抖动1年余”于2016年8月17日入院。患儿自幼精神、运动落后于同龄儿童,动作笨拙,至今走路不稳,有时会跌倒,出牙迟,牙齿未出全,语言发育比同龄儿落后。1年余前患儿无明显诱因出现双手抖动,持物、取物时手抖动明显,无四肢乏力,无吞咽困难,无呛咳。未予诊治,上述症状持续未改善。为进一步诊治前来我院。患儿无兄弟姐妹,否认类似疾病家族史。查体:体质量24 kg,身高121.9 cm,神志清,反应稍迟钝,语言欠清晰,面容略显幼稚,双侧瞳孔等大等圆,对光反射存在,眼球震颤,咽反射正常,牙列不齐,出牙不全,颈软,心、肺、腹部未及异常,行走不稳,宽基底步态,意向性震颤,四肢肌力Ⅴ级,双下肢肌张力稍增高,角膜反射、腹壁反射正常,膝反射、跟腱反射活跃,痛觉存在(其他感觉检查不合作),双侧Kernig征、Brudzirnski征、Babinski征阴性。头颅MRI检查示两侧大脑半球对称,灰白质分界不清,双侧大脑半球脑白质呈弥漫性长T2信号,Flair及DWI稍高信号。入院后行血尿便常规、肝肾功能、电解质、甲状腺功能、促肾上腺皮质激素检查未见异常,皮质醇157.5 nmol/L低于正常(正常范围171~536 nmol/L)。视频ECG正常。四肢神经传导及EMG未见明显异常。视觉诱发电位双侧P100潜伏期延长,听觉诱发电位双侧Ⅰ波潜伏期延长、双侧ABR主观听阈轻度增高。智商49分,中度弱智。眼科检查示近视、视神经萎缩。入院后临床诊断为脑白质营养不良?送基因检验,北京康旭医学检验所报告(二代测序,一代验证)示患儿POLR3A复合杂合核苷酸变异:POLR3A(NM_007055.3):c.1781T>G(p.Leu594Arg)编码区第1781号核苷酸由T变为G的杂合核苷酸变异,该变异导致第594号氨基酸由Leu变为Arg,为错义突变(图1),为可能致病突变;POLR3A(NM_007055.3):c.2693delT(p.Ile898 ThrfsTer10),编码区第2693号核苷酸T缺失的杂合核苷酸变异,该变异导致从第898号氨基酸Ile开始的氨基酸合成发生改变,并在改变后的第10个氨基酸终止,为移码突变(图2)。蛋白质功能损伤预测:SIFT提示有可能致病,PolyPhen2提示很可能致病,MutationTaster提示很可能致病。上述变异均可能导致蛋白质功能受到影响。受检者上述变异分别遗传自其父母,其父母均只携带其中一个杂合变异。明确诊断为4H综合征。门诊随访至截稿,患儿不规则口服维生素B1、维生素B12、赖氨酸等药物,患儿认知、运动、步态情况未有改善,左侧上中切牙长出,右侧上中切牙萌出。
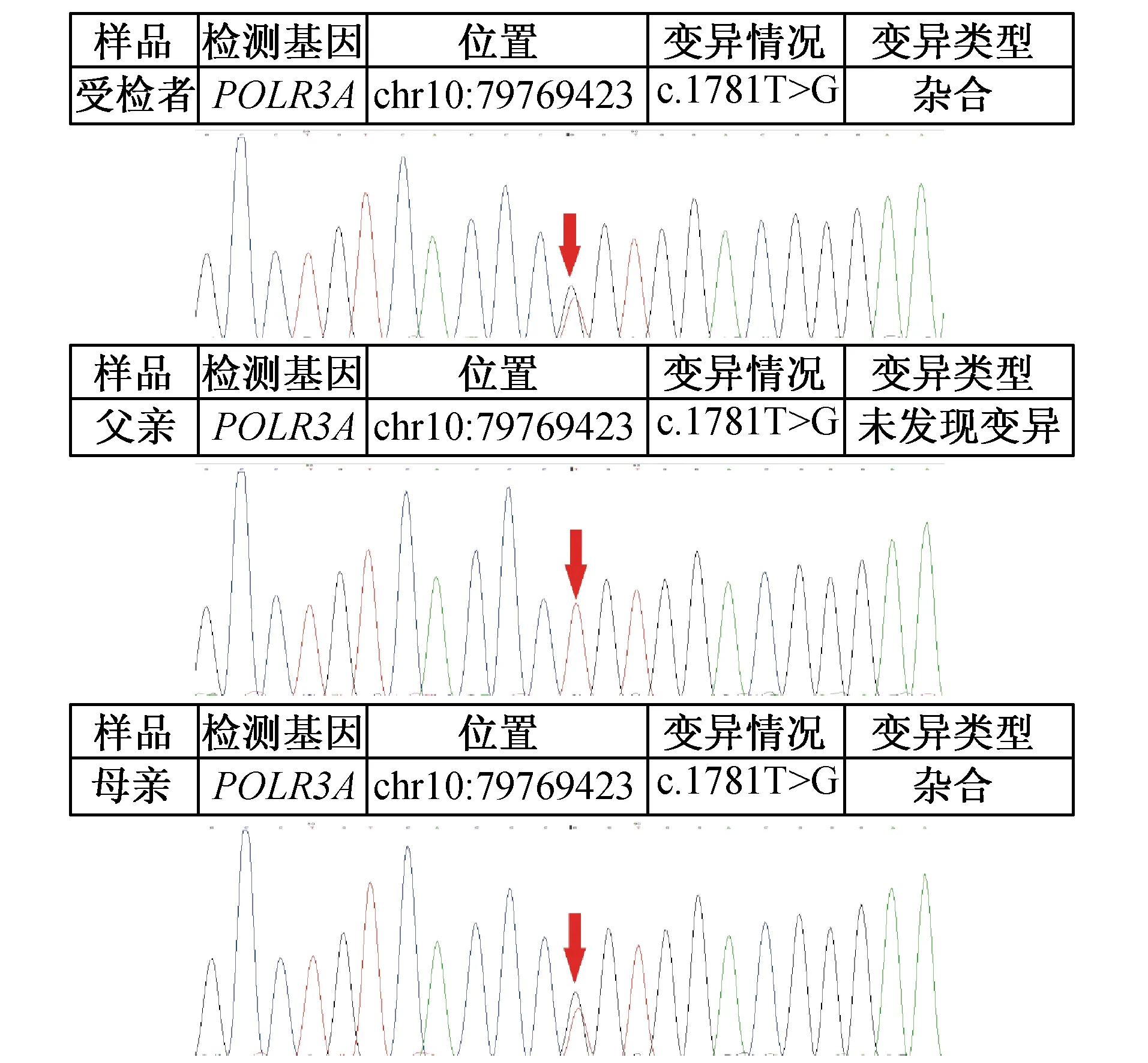
图1 患儿及其母亲POLR3A基因c.1781T>G错义突变
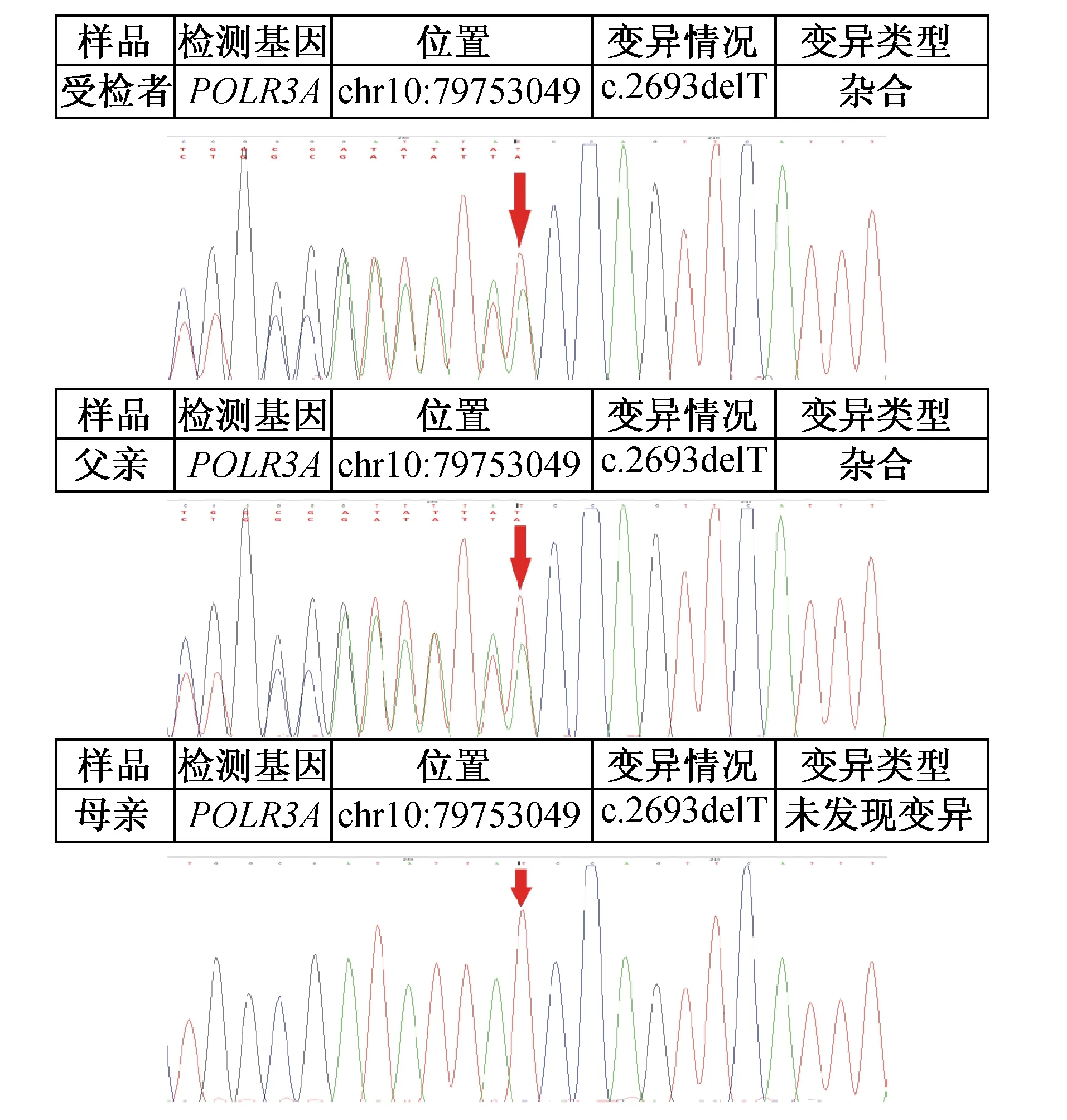
图2 患儿及其父亲POLR3A基因c.2693delT移码突变
2 讨 论
2005年Wolf等[1]报道4例共济失调、出牙延迟、脑白质髓鞘化不良(ADDH)的患者;2006年Timmons等[2]报道4例表现为脑白质髓鞘化不良、低促性腺激素性性腺功能减退、少牙畸形的患者,首先提到4H综合征;2011年Sato等[3]报道ADDH与4H综合征有重叠表现;2013年Takanashi等[4]、2015年Muthusamy等[5]报道中提到除有上述改变外,还可以有白内障、胼胝体及小脑萎缩、椎体融合、周围神经髓鞘形成不良等表现,包含髓鞘发育不良性脑白质营养不良7型(HLD7)等患者应属于4H综合征谱系或POLⅢ相关脑白质营养不良。
2008年Wolf等[6]、2012年Terao等[7]、2013年Vanderver等[8]报道的4H综合征中多有运动、语言发育落后,出牙延迟,小脑共济失调、脑白质髓鞘化不良、低促性腺激素性性腺功能减退等临床症状,随访发现低睾酮、低生长激素、低皮质醇水平以及腰椎骨密度降低、身高低于同龄儿。上述报道均说明4H综合征临床表现出现早晚存在明显的个体差异,总结并掌握其核心特征有助于早期诊断、对症治疗,避免过多检查,减轻家庭经济负担。本文患儿精神、运动发育落后、共济失调、出牙延迟、脑白质病变与Vanderver等[8]报道的患儿早期表现类似,说明精神、运动发育迟缓、动作笨拙是4H综合征婴幼儿期需要注意的表现,此时发现出牙延迟及头颅MRI脑白质病变可以考虑4H综合征,进行相关基因检查。Potic等[9]报道4H综合征患者在20岁常规激素筛查发现生长激素缺乏,但没有身材矮小,提出不需要常规评价生长激素刺激试验,在伴有其他内分泌激素缺乏表现时,可进行生长激素、性激素、皮质醇测定。Terao等[7]、Wolf等[10]报道50%4H综合征出现身材矮小,牙齿和激素的变化并不总是出现,可能说明身材矮小、牙齿和激素的变化只是4H综合征的普通表现,并不作为核心症状。本文患儿诊断年龄早,就诊时有出牙延迟,并未出现身材矮小,未到性征发育年龄,除测定皮质醇略低于正常水平外,内分泌科会诊后未进行生长激素及性激素测定,建议其密切随访。另外本文患儿虽未主观反映视力、听力问题,在眼科及听力检查中发现近视、视神经萎缩、视觉诱发电位双侧P100潜伏期延长,听觉诱发电位双侧Ⅰ波潜伏期延长、双侧ABR主观听阈轻度增高, 与Wolf等[10]报道的几乎所有患者会出现近视相一致,视、听检查异常与视辐射、视、听神经髓鞘化不良有关。本文患儿头颅MRI检查未发现小脑萎缩及胼胝体变薄,与Wolf等[10]报道POLR3A 突变患者中,小脑萎缩通常不会出现或者很轻微,胼胝体在年纪小的儿童中通常是正常的,在成人中永远是变薄的相一致。
2011年Bernard等[11]报道4H综合征为POLR3A核苷酸变异引起;2013年Daoud等[12]报道POLR3A或POLR3B核苷酸变异均可引起4H综合征,并提出POLR3A核苷酸突变更常见、更频繁;2014年Wolf等[10]提出POLR3A基因核苷酸突变引起的4H综合征病情更严重。在既往研究[10-12]中,4H综合征患者POLR3A或POLR3B基因不同核苷酸位点变异(如c.3013C>T、c.2011T>C、c.1302insA、c.2084-6A>G等)被报道,并不断有新的突变位点被发现。本文患儿POLR3A复合杂合核苷酸变异:c.1781T>G,c.2693delT,尚未见文献报道过(所参考数据库:HGMDPro及PubMed),为新发现突变位点。
综上所述,本文报道了1例早期基因诊断的4H综合征患儿,并结合文献资料,总结出精神、运动发育迟缓、出牙延迟可能是4H综合征早期表现,近视、共济失调、脑白质髓鞘化不良可能是其核心特征,部分患者可出现低促性腺激素性性腺功能减退、少牙畸形等改变,基因检查提示POLR3A或POLR3B突变。
[1] Wolf NI, Harting I, Boltshauser E, et al. Leukoencephalopathy with ataxia, hypodontia, and hypomyelination[J]. Neurology, 2005, 64: 1461.
[2] Timmons M, Tsokos M, Asab MA, et al. Peripheral and central hypomyelination with hypogonadotropic hypogonadism and hypodontia[J]. Neurology, 2006, 67: 2066.
[3] Sato I, Onuma A, Goto N, et al. A case with central and peripheral hypomyelination with hypogonadotropic hypogonadism and hypodontia (4H syndrome) plus cataract[J]. J Neurol Sci, 2011, 300: 179.
[4] Takanashi J, Osaka H, Saitsu H, et al. Different patterns of cerebellar abnormality and hypomyelination between POLR3A and POLR3B mutations[J]. Brain Development, 2013, 36: 259.
[5] Muthusamy K, Sudhakar SV, Yoganathan S, et al. Hypomyelination, hypodontia, hypogonadotropic hypogonadism (4H) syndrome with vertebral anomalies: a novel association[J]. J Child Neurol, 2015, 30: 937.
[6] Wolf NI, Harting I, Innes AM, et al. Ataxia, delayed dentition and hypomyelination: a novel leukoencephalopathy[J]. Neuropediatrics, 2007, 38: 64.
[7] Terao Y, Saitsu H, Segawa M, et al. Diffuse central hypomyelination presenting as 4H syndrome caused by compound heterozygous mutations in POLR3A, encoding the catalytic subunit of polymerase Ⅲ[J]. J Neurol Sci, 2012, 320: 102.
[8] Vanderver A, Tonduti D, Bernard G, et al. More Than Hypomyelination in Pol-Ⅲ Disorder[J]. J Neuropathol Expe Neurol, 2013, 72: 67.
[9] Potic A, Brais B, Choquet K, et al. 4H syndrome with late-onset growth hormone deficiency caused by POLR3A mutations[J]. Arch Neurol, 2012, 69: 920.
[10] Wolf NI, Vanderver A, van Spaendonk RM, et al. Clinical spectrum of 4H leukodystrophy caused by POLR3A and POLR3B mutations[J]. Neurology, 2014, 83: 1898.
[11] Bernard G, Chouery E, Putorti ML, et al. Mutations of POLR3A encoding a catalytic subunit of RNA polymerase PolⅢ cause a recessive hypomyelinating leukodystrophy[J]. Am J Hum Genet, 2011, 89: 415.
[12] Daoud H, Tétreault M, Gibson W, et al. Mutations in POLR3A and POLR3B are a major cause of hypomyelinating leukodystrophies with or without dental abnormalities and/or hypogonadotropic hypogonadism[J]. J Med Genet, 2013, 50: 194.
Clinicalfeaturesandmoleculardiagnosisof4Hsyndrome(reportof1case)
LIANGChao,LUHai-ying,GUOHu.
DepartmentofNeurology,Children’sHospitalofNanjingMedicalUniversity,Nanjing210008,China
ObjectiveTo explore the clinical characteristics and molecular diagnosis of 4H syndrome.MethodsThe clinical data of 1 patient with 4H syndrome diagnosed by gene was retrospectively analyzed, and the clinical characteristics were analyzed combined with the literature.ResultsThis child patient was male, 6 years and 8 months old, with hands shake for 1 years, mental and movement development backwardness, walking instability, teething delay. Ophthalmic examination showed myopia and optic atrophy. Brain MRI suggested a wide range of cerebral white matter lesions on both sides of the cerebral hemisphere. Gene examination showed POLR3A compound heterozygous mutation (c.1781T>G,c.2693delT). He was diagnosed as 4H syndrome.ConclusionsThe early manifestations of 4H syndrome are mental and movement development backwardness and teething delay. The main clinical features of 4H syndrome are leukodystrophy, myopia and ataxia. The genetic characteristics are POLR3A or POLR3B mutation.
4H syndrome;clinical feature;genetic feature
R748
A
1004-1648(2017)05-0377-03
南京市科技发展计划项目(201402021)
210008南京医科大学附属儿童医院神经内科
陆海英,郭虎
2016-11-28
2016-12-23)
