3,3′ -双(2,2-二甲基-5-硝基-1,3-二氧杂环己烷-5-ONN-氧化偶氮基)氧化偶氮呋咱(BDDAF)的合成、晶体结构与热行为
2017-11-01张家荣毕福强王伯周张俊林贾思媛
张家荣,毕福强,王伯周,张俊林,贾思媛
(1.氟氮化工资源高效开发与利用国家重点实验室,陕西 西安 710065;2.西安近代化学研究所,陕西 西安 710065)
3,3′ -双(2,2-二甲基-5-硝基-1,3-二氧杂环己烷-5-ONN-氧化偶氮基)氧化偶氮呋咱(BDDAF)的合成、晶体结构与热行为
张家荣1,2,毕福强1,2,王伯周1,2,张俊林1,2,贾思媛1,2
(1.氟氮化工资源高效开发与利用国家重点实验室,陕西 西安 710065;2.西安近代化学研究所,陕西 西安 710065)
以三羟甲基硝基甲烷(TN)和丙酮为原料,通过缩合、碱催化、亚硝化反应合成了2,2-二甲基-5-硝基-5-亚硝基-1,3-二氧杂环己烷(DMNNDO),然后以DMNNDO与二氨基氧化偶氮呋咱(DAOAF)为原料, 经氧化偶联反应合成了含能中间体3,3′ -双(2,2-二甲基-5-硝基-1,3-二氧杂环己烷-5-ONN-氧化偶氮基)氧化偶氮呋咱(BDDAF),4步反应总收率为40%;采用IR、1H NMR、13C NMR、15N NMR和元素分析对目标化合物进行了结构表征;培养并获得了BDDAF的单晶,利用X-射线单晶衍射仪对其结构进行了表征;利用差示扫描量热(DSC)法和热重分析(TG)法研究了BDDAF的热行为。结果表明,BDDAF为单斜晶系,空间群为P2(1)/n,晶胞参数为a=0.54504(15) nm,b=3.1462(9),c=0.7546(2),β=103.612(5)°,V=1.2577(6)nm3,Z=2,Dc=1.554g/cm3,F(000)=608,wR1=0.1387,wR2=0.3306;优化了氧化偶联反应条件,收率达到85%;改进了氧化偶联的后处理方法,采用溶剂-非溶剂法代替柱层析分离法,产品纯度大于99%;BDDAF的熔点为117℃,热分解温度为248℃。
有机化学;三羟甲基硝基甲烷;氧化偶氮呋咱;含能化合物;BDDAF
引 言
能量和密度是发展新型含能化合物的两项重要指标[1-4]。多硝甲基氧化偶氮基作为一种高能密度含能基团,近年来受到广泛关注。由于此基团通过碳原子将多个硝基和氧化偶氮基结合组成新型高密度基团,若将其引入现有含能母体结构,可以有效提高含能化合物的能量和密度。俄罗斯泽林斯基研究院对此含能基团进行了多年的研究[2-15],将该基团引入氮杂含能母体结构,如呋咱环、呋咱醚和氧化呋咱环等,合成了多种性能优良的含能化合物。呋咱环和氧化偶氮基中含有大量N-N和C-N键,使含能化合物分解时放出大量氮气,产生较高能量[16]。西安近代化学研究所[17]对此类含能化合物的爆轰性能进行了理论模拟计算,如3,3′-双[α-(二硝甲基)氧化偶氮基]氧化偶氮呋咱、3,3′-双[α-(三硝甲基)氧化偶氮基]氧化偶氮呋咱、3,3′-双[α-(氟二硝甲基)氧化偶氮基]氧化偶氮呋咱等,发现该类化合物均具有良好的爆轰性能,合成上述含能化合物,首先需要通过伯胺的氨基和DMNNDO中的亚硝基发生氧化偶联反应,在现有含能母体结构上引入多硝甲基烷基氧化偶氮基,而后发生一系列的官能团转化,从而可以合成多硝甲基烷基氧化偶氮含能化合物[18],如何提高该氧化偶联步骤反应收率,确定多硝甲基氧化偶氮基的微观构象,是进一步合成此类化合物的基础。
本实验以三羟甲基硝基甲烷(TN)为原料,通过氧化偶联反应合成了含能中间体3,3′-双(2,2-二甲基-5-硝基-1,3-二氧杂环己烷-5-ONN-氧化偶氮基)氧化偶氮呋咱(BDDAF),并对其结构进行了表征;筛选了氧化偶联步骤的反应溶剂,考察了反应料比、反应温度等因素对氧化偶联反应的影响;采用溶剂-非溶剂法代替传统柱色谱分离法,改进了氧化偶联步骤的后处理方法,有效提高化合物的纯度;采用DSC-TG研究了BDDAF的热行为,为进一步探索此类结构的含能化合物奠定基础。
1 实 验
1.1 试剂与仪器
DAOAF参考文献[19]制备;三羟甲基硝基甲烷(TN)、丙酮、对甲苯磺酸(TsOH)、甲醇钠的甲醇溶液(质量分数30%)、CH3CH2OH、四氧化二氮、CH2Cl2、1,3-二溴-1,3,5-三嗪-2,4,6-三酮(DBI) (纯度为98%)均为市售分析纯试剂。
NEXUS 870 型傅里叶变换红外光谱仪,美国 Nicolet公司;AV 500 型(500MHz) 超导核磁共振仪、AVANCE Ⅲ HD (800MHz) 超导核磁共振仪,瑞士Bruker公司;Vario EL Ⅲ 型自动微量有机元素分析仪,德国Elementar gongsi公司;DSC-204差示扫描热量仪,STA449C型热重仪,德国Netzsch公司。
1.2 目标化合物的合成路线
目标化合物的合成路线如下:
1.2.1 2,2-二甲基-5-硝基-5-羟甲基-1,3-二氧杂环己烷 (DHND)的合成
将0.8mol TN(120.8g)和13.4mmol TsOH (2.3g)加入176.4mL丙酮中,反应液加热至40℃直至固体全部溶解,加入无水硫酸镁,升温至60℃反应24h,降至室温,将反应液倒入冰水中搅拌30min,过滤,水洗,晾干,得到134.5g浅黄色固体DHND,收率为88%。
1H NMR(DMSO-d6,500 MHz),δ: 1.252 (s, 3 H, CH3), 1.401 (s, 3 H, CH3), 3.728 (d, 2H,J=5.7Hz, CH2), 4.051(d,J=13Hz, 2H, CH2), 4.350 (d,J=13Hz, 2 H, CH2), 5.473 (t,J=5.7Hz, 1H, OH);13C NMR(DMSO-d6, 125MHz),δ: 20.591 (CH3), 27.214 (CH3), 61.604 (CH2O), 62.898 (-CH2-OH), 87.922 (CNO2), 98.891 (OCO); IR(KBr),ν(cm-1): 3427 (OH), 2949, 1386 (CH3), 2894 (CH2), 1542,1352 (NO2), 1061 (CO); 元素分析(C7H13O5N, %): 理论值, C 43.98; H 6.85; N 7.33; 实测值, C 42.69 , H 6.705 ; N 7.159。
1.2.2 2,2-二甲基-5-硝基-5-亚硝基-1,3-二氧杂环己烷(DMNNDO)的合成
将7.3mmol DHND(1.4g)加入20mL无水乙醇中,滴入14.6mmol甲醇钠的甲醇溶液(2.64g),升温至70℃,反应完毕,降至室温,然后减压抽去反应体系中易挥发物质,加入200mL CH2Cl2,将温度降至-35℃,然后滴入74mmol N2O4(6.8g)的CH2Cl2溶液。30min后,将温度升至0~5℃,过滤。将滤液浓缩,通过硅胶层,用CH2Cl2洗脱,浓缩有机相,将得到的固体冷冻30min,用乙醚洗涤得到0.75g DMNNDO,收率为54%。
1H NMR (CDCl3-d6, 800MHz),δ: 1.4567 (s, 3 H, CH3), 1.5555 (s, 3 H, CH3), 4.4228 (d,J=13.54, 2 H, CH2), 4.5401 (d,J=13.44, 2 H, CH2);13C NMR (CDCl3-d6, 200MHz),δ: 22.1173 (CH3), 24.1947 (CH3), 58.6353 (CH2O), 100.3268 (OCO), 130.7206 (CNO2). IR (KBr),ν(cm-1): 2989, 2957, 1379, 1687, 1584, 1449, 1360, 1379, 1327; 元素分析(C6H10O5N2, %): 理论值, C 37.89, H 5.263, N 14.73; 实测值, C 37.67, H 5.242, N 13.63。
1.2.3 3,3′-双(2,2-二甲基-5-硝基-1,3-二氧杂环己烷-5-ONN-氧化偶氮基)氧化偶氮呋咱(BDDAF)的合成
20℃下,向250mL 三口烧瓶中加入10mmol(1.9g)DMNNDO和65mL二氯甲烷,搅拌溶解后,加入4.16mmol(0.88g)二氨基氧化偶氮呋咱(DAOAF)和15mmol(4.3g)1,3-二溴-1,3,5-三嗪-2,4,6-三酮(DBI),在此温度下搅拌。利用TLC薄层色谱监测反应,直至原料分解完毕(约13h)。室温下减压蒸出二氯甲烷,通过硅胶色谱柱,以石油醚和乙酸乙酯体积比为8∶1的溶剂进行洗脱分离,减压蒸馏除去溶剂,晾干得 2g 浅黄色固体,收率为85%。
1H NMR(CDCl3-d6,500MHz)δ: 1.484 (s, 3 H, CH3), 1.496 (d, 6 H,J=2.5, CH3), 1.519 (s, 3 H, CH3), 4.769~4.817(m, 8 H, CH2);13C NMR(CDCl3-d6,125MHz)δ: 21.735, 22.102, 23.780, 24.102 (CH3), 62.335, 62.524 (CH2), 101.090, 101.193, 107.826, 108,132(C), 148.520, 149.614, 149.650, 153.565 (furazan cycles);15N NMR (CDCl3-d6, 125MHz)δ:41.297, 36.755, 36.651, -13.075, -13.986, -41.743,-46.041, -58.892, -65.484, -66.155, -75.360; IR (KBr),ν(cm-1): 3000, 2947, 2917, 1583, 1503, 1380, 1201; 元素分析(C16H20N12O13, %): 理论值, C 32.65, H 3.4, N 28.57; 实测值, C 32.57, H 3.303, N 28.22。
1.3 单晶培养
培养并获得了关键中间体BDDAF的单晶,通过X-射线单晶衍射仪测试了其晶体结构。得到BDDAF晶体属于单斜晶系,空间群为P2(1)/n,一个晶胞内含有2个分子。
1.4 热行为研究
采用差式扫描量热仪测试了目标化合物的热性能。测试条件为:动态氮气气氛、温度范围10~500℃,压力0.1MPa,升温速率10℃/min,试样皿为铝盘。
2 结果与讨论
2.1 BDDAF合成条件的优化
2.1.1 氧化偶联反应溶剂的选择
以二氯甲烷为反应介质,原料DAOAF和氧化剂DBI在二氯甲烷中溶解性较差,需较长时间完成反应,因而尝试采用DMSO作为反应溶剂。原料DAOAF和DMNNDO虽然都可以在DMSO中溶解,但DBI仍不能完全溶解,利用薄层色谱板检测反应,发现12h后无产物生成, 因此DMSO并不是反应的最佳介质。这可能是因为原料DMNNDO在DMSO中不能长时间稳定存在而导致反应无法进行,因此采用对DMNNDO较稳定的二氯甲烷为溶剂进行反应,得到BDDAF的收率为85%。
2.1.2 料比对氧化偶联反应收率的影响
氧化偶联反应是一个双官能团反应,因此反应的料比也是影响收率的关键因素。在反应温度20℃、反应时间为12h条件下,考察了不同料比(DAOAF、DMNNDO与DBI的摩尔比)对BDDAF收率的影响,结果如表1所示。
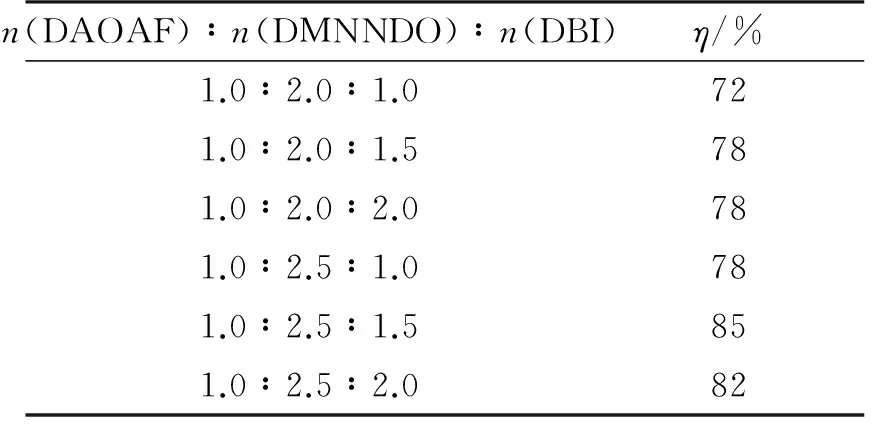
表1 氧化偶联反应料比对BDDAF收率的影响
由表1可以看出,当DAOAF、DMNNDO与DBI的摩尔比为1.0∶2.0∶1.0时,得到的产物经核磁共振鉴定有目标化合物BDDAF的存在,经过纯化处理,收率为72%。这可能是因为该步为双官能团反应,DMNNDO需稍过量才能保证两边基团都反应完全。随着DMNNDO加入量增多,氧化偶联反应的收率逐渐增高,当DAOAF、DMNNDO和DBI的摩尔比为1.0∶2.5 ∶1.5时,收率达85%,继续增大DMNNDO比例,收率变化不大,因此,该步反应最佳反应料比为1.0∶2.5∶1.5。
2.1.3 温度对氧化偶联反应收率的影响
在DAOAF与DMNNDO摩尔比为1.0∶2.5,反应时间12 h的情况下,分别考察了反应温度为0、10、20和30℃时对BDDAF收率的影响,得到BDDAF收率分别为53%、68%、85%和76%。
在0~20℃内,随着反应温度升高,BDDAF的收率逐渐升高。当温度为20℃时,反应收率最高达到85%,继续升高反应温度则收率略有下降。分析认为温度升高至30℃时,达到了溶剂二氯甲烷的沸点,部分二氯甲烷挥发,导致收率下降。因此,综合考虑,反应温度20℃为宜。
2.2 BDDAF单晶结构解析
BDDAF晶体的分子结构和晶胞堆积图见图1,晶体结构数据及精修参数、主要键长、键角及氢键数据见表2~表5。
从图1(a)和表3可以看出,BDDAF中,两个二氧六环均为椅式构象。六元环中,6个原子分别处在椅式一上一下的位置,向下的碳原子C3、C4和C6组成的平面和向上的原子O1、O2和C6组成的平面相互平行。其中C4-C6和C5-C6的键长分别为0.151和1.52nm,与正常的C-C的键长(0.153nm)相符;而六元环中4个碳氧键C3-O1、C3-O2、C4-O1、C5-O2的键长分别为0.142、0.143、 0.141和0.140nm,与常规醚类化合物中的碳氧键长(0.142nm)相符;六元环中C-C键长和C-O键长未受到成环及周围基团的影响,表明这种构象没有因为键长变形而引起内能升高,也没有因为存在角张力使得整个分子的势能降低,分子更加稳定。而六元环中与C2相连的两个甲基和与C6相连的硝基和氧化偶氮基团都分别处于直立键与平伏键,C2-N12两个原子之间的距离为0.442nm,C1-N11原子间距离为0.457nm,都远大于两个原子半径之和,取代基间没有排斥力,因此,分子中两个六元环的构型是一个低能量的稳定构象。

表2 BDDAF的晶体数据和结构精修参数
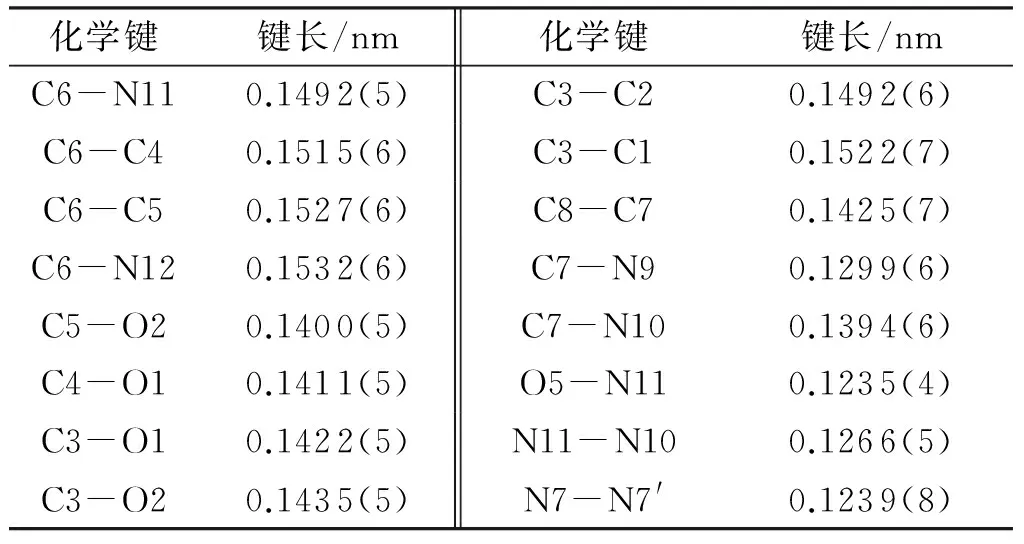
表3 BDDAF的部分键长
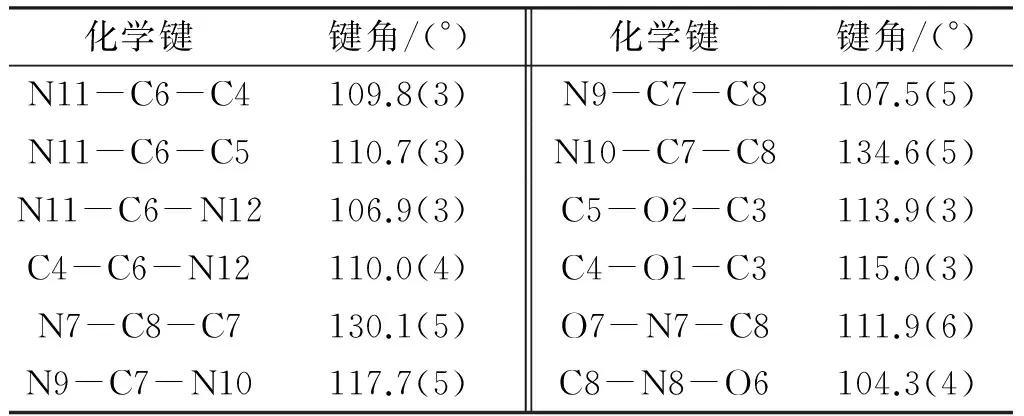
表4 BDDAF的部分键角

表5 BDDAF氢键的键长和键角
由表3可看出,BDDAF晶体中,N10=N11的键长为0.126nm,比N7=N7′的键长(0.123nm)长,N11-O5的键长为0.123nm,比N7=O7′键长(0.124nm)短,表明这两类氧化偶氮键有细微差别,和常规N=N(0.124nm) 与N=O(0.121nm)相比较为相似,故晶体中的氧化偶氮键键长并没有因为特殊结构发生明显变化。晶体中C7-N10和C8-N7的键长分别为0.139nm和0.141nm,而C6-N11和C6-N12的键长分别为0.1490和0.1532nm,说明氧化偶氮呋咱环和与其相连的两个氧化偶氮基共轭,从而使化合物更加稳定。
从图1(b)和表5可以看出,BDDAF分子间还存在两种氢键。两层分子间C1-H1C…O3的键长为0.330nm,键角为147.1°,两列分子间C1-H1C…O5的键长为0.361nm,键角为149.5°,均为典型的较强的分子间氢键,促使BDDAF分子形成紧凑稳定的晶体结构。
2.3 BDDAF的热行为
采用差示扫描量热法(DSC)和热重分析法(TG)对BDDAF的热分解行为进行了研究,结果见图2。
由图2可见,DSC曲线在117℃有一个吸热峰,为BDDAF的融化峰,在248℃的放热峰为该化合物的分解峰,表明BDDAF先经历吸热融化的相变过程,然后再进行分解。从TG曲线来看,在181℃之前,该物质失重很小,仅为1.5%,说明当温度低于181℃时,该物质比较稳定;在温度从181℃升至263℃的过程中,BDDAF快速失重,累计分解深度为75.9%。随着温度的升高,BDDAF进一步分解,在443℃时,BDDAF基本分解完全,残留9.3%,剩余残渣少。
3 结 论
(1)以三羟甲基硝基甲烷(TN)和丙酮为原料,经过缩合、碱催化、亚硝化合成了DMNNDO,利用DMNNDO和DAOAF的氧化偶联反应,合成了BDDAF,总收率为40%。
(2)培养并获得了BDDAF的单晶,通过晶体结构解析,发现BDDAF中的六元环呈稳定的椅式构型,引入的氧化偶氮结构为反式构型;氧化偶氮基和呋咱环共轭,并且存在两种分子间氢键,有效提高了化合物的稳定性。
(3)BDDAF的熔点为117℃,热分解峰温为248℃,热稳定性较好。
[1] 张俊林,肖川,翟连杰,等. 多硝基氮杂稠环化合物合成及性能[J]. 有机化学, 2016, 36(6): 1197-1207.
ZHANG Jun-lin, XIAO Chuan, ZHAI Lian-jie, et al. Synthesis and properties of the fused aza-polynitrocyclic compounds[J]. Chinese Journal of Organic Chemistry, 2016, 36(6): 1197-1207.
[2] 张敏,毕福强,许诚,等. 双[2-(5-硝基-2H-四唑基)-2-2-二硝乙基]-硝胺的合成与量子化学计算[J]. 火炸药学报, 2014, 37(5): 52-57.
ZHANG Min, BI Fu-qiang, XU Chen, et al. Synthesis and quantum chemistry calculation of bis[2- (5-nitrotetrazol-2-yl)-2,2-dinitroethyl]nitroamine[J]. Chinese Journal of Explosives & Propellants(Huozhayao Xuebao), 2014, 37(5): 52-57.
[3] 王晓峰. 军用混合炸药的发展趋势[J]. 火炸药学报, 2011, 34(4):1-4.
WANG Xiao-feng. Developmental trends in military composite explosive[J]. Chinese Journal of Explosives & Propellants(Huozhayao Xuebao), 2011, 34(4):1-4.
[4] 魏伦,王琼林,刘少武,等. 高能量密度化合物CL-20、DNTF和ADN 在高能发射药中的应用[J]. 火炸药学报, 2009, 32(1): 17-20.
WEI Lun, WANG Qiong-lin, LIU Shao-wu, et al. Application of high energy density compounds CL-20,DNTF and ADN in high energy propellant[J]. Chinese Journal of Explosives & Propellants(Huozhayao Xuebao), 2009, 32(1): 17-20.
[5] Luk′yanov O A, Pokhvisneva G V, Ternikova T V. Bis (nitro-and polynitromethyl-ONN-azoxy) azoxyfurazans and some of their derivatives[J]. Russian Chemical Bulletin, 2012, 61(9): 1783-1786.
[6] Luk′yanov O A, Salamonov Y B, Bass A G, et al. Aryl-NNO-azoxy-α-nitroalkanes[J]. Bulletin of the Russian Academy of Sciences, Division of Chemical Science, 1992, 41(10): 1884-1894.
[7] Luk′yanov O A, Pokhvisneva G V, Ternikova T V, et al. Aliphatic α-nitroalkyl-ONN-azoxy compounds and their derivatives[J]. Russian Chemical Bulletin, 2009, 58(10): 2063-2069.
[8] Luk′yanov O A, Pokhvisneva G V, Ternikova T V, et al. α-Nitroalkyl-ONN-azoxyfurazanes and some of their derivatives[J]. Russian Chemical Bulletin, 2011, 60(8): 1703-1711.
[9] Luk′yanov O A, Pokhvisneva G V, Ternikova T V, et al. 3, 4-Bis (α-nitroalkyl-ONN-azoxy) furazans and some of their derivatives[J]. Russian Chemical Bulletin, 2012, 61(2): 360-365.
[10] Luk′yanov O A, Parakhin V V, Pokhvisneva G V, et al. 3-Amino-4-(α-nitroalkyl-ONN-azoxy) furazans and some of their derivatives[J]. Russian Chemical Bulletin, 2012, 61(2): 355-359.
[11] Luk′yanov O A, Parakhin V V. 3-(α-Nitroalkyl-and α-polynitroalkyl-ONN-azoxy)-4-nitraminofurazans and some their derivatives[J]. Russian Chemical Bulletin, 2012, 61(8): 1582-1590.
[12] Gottardi W. über Bromierungen mit dibromoisocyanurs?ure unter ionischen Bedingungen, 1. Mitt.:Monobromierungen [J]. Monatshefte für Chemie / Chemical Monthly, 1968, 99(2): 815-822.
[13] Luk′yanov O A, Pokhvisneva G V, Ternikova T V. Nitro-substituted bis(methyl-ONN-azoxyfurazanyl)furoxans[J]. Russian Chemical Bulletin, 2015, 64(1): 137-141.
[14] Luk′yanov O A, Salamonov B Yu, Bass G A, et al. N′-(α-acetoximinoalkyl) diazene-N-oxides and some of their transformations[J]. Bulletin of the Academy of Sciences of the USSR, Division of Chemical Science, 1991, 40(1): 93-98.
[15] Luk′yanov O A, Salamonov B Yu, Bass G. A, et al. Aryl-NNO-azoxy-of- nitroalkanes [J]. Izvestiya Akademi Nauk, Seriya Khimicheskaya, 1992, 10: 2400-2412.
[16] 翟连杰,王伯周,霍欢,等. 3-氨基-4-偕羟胺肟基呋咱合成、歧化与水解反应研究[J]. 有机化学, 2013, 33(8), 1755-1761.
ZHAI Lian-jie, WANG Bo-zhou, HUO Huan, et al. Synthesis, disproportionation and hydrolysis reactions of 3-amino-4-hydroxyaminoximinofurazan[J]. Chinese Journal of Organic Chemistry, 2013, 33(8), 1755-1761.
[17] 毕福强, 王玉, 王伯周, 等. 多硝甲基氧化偶氮呋咱含能衍生物爆轰与安全性能理论研究[J].含能材料, 2016, 24(11):1063-1069.
BI Fu-qiang, WANG Yu, WANG Bo-zhou, et al. Theoretically study on the detonation and safety properties of energetic derivatives based on polynitromethylazoxyfurazan[J]. Chinese Journal of Energetic Materials, 2016, 24(11):1063-1069.
[18] 张家荣, 毕福强, 王伯周. 等. α-多硝甲基氧化偶氮含能化合物合成研究进展[J]. 火炸药学报, 2016,39(6):12-19.
ZHANG Jia-rong, BI Fu-qiang, WANG Bo-zhou, et al. Research progress on the synthesis of energetic compounds based on α-polynitromethyl-azoxy moieties[J]. Chinese Journal of Explosives & Propellants(Huozhayao Xuebao), 2016,39(6):12-19.
[19] 李洪珍,黄明,李金山,等. 3,3′-二氨基-4,4′-偶氮呋咱及其氧化偶氮呋咱的合成[J]. 含能材料, 2004,12(zl): 79-81.
LI Hong-zhen, HUANG Min, LI Jin-shan, et al. Synthesis of diaminoazofurazan and diaminoazoxyfurazan[J]. Chinese Journal of Energetic Materials, 2004,12(zl): 79-81.
Synthesis,CrystalStructureandThermalBehaviorof3,3′-Bis(2,2-dimethyl-5-nitro-1,3-dioxan-5-yl-ONN-azoxy)azoxyfurazan(BDDAF)
ZHANG Jia-rong1,2, BI Fu-qiang1,2, WANG Bo-zhou1,2, ZHANG Jun-lin1,2, JIA Si-yuan1,2
(1.State Key Laboratory of Fluorine & Nitrogen Chemical, Xi′an 710065, China; 2. Xi′an Mordern Chemistry Research Institute, Xi′an 710065, China)
Taking tri(hydroxymethyl)nitromethane(TN) and acetone as raw materials, 2,2-dimethyl-5-nitro-5-nitroso-1,3-dioxane(DMNNDO) was synthesized via. condensation, alkaline hydrolysis and nitrosation reactions. The energetic intermediate 3,3′-bis(2,2-dimethyl-5-nitro-1,3-dioxan-5-yl-ONN-azoxy)azoxyfurazan(BDDAF) was synthesized via. oxidative-coupling reaction taking DMNNDO and DAOAF as starting materials, and the total yield of this four-step reactions was 40%. The structure of target compound was characterized by IR,1H NMR,13C NMR,15N NMR and elemental analysis. The single crystal of BDDAF was cultured and its structure was characterized by X-ray single crystal diffractometer. The thermal behavior of BDDAF was investigated by differential scanning calorimetry(DSC) and thermogravimetric analysis (TG) method. The results show that the crystal belongs to monoclinic system, space group isP2(1)/n, cell parameters area=0.54504(15)nm,b=3.1462(9),c=0.7546(2),β=103.612(5)°,V=1.2577(6)nm3,Z=2,Dc=1.554g/cm3,F(000)=608,wR1=0.1387 andwR2=0.3306. The oxidative coupling reaction conditions are optimized and the yield can reach 85%.The solvent-nonsolvent method is used instead of column chromatography, the purity of BDDAF is more than 99%, and the melting point and decomposition temperature are 117℃ and 248℃,respectively.
organic chemistry; tri(hydroxymethyl)nitromethane; azoxy frazan; energetic compounds; BDDAF
TJ55;O62
A
1007-7812(2017)05-0045-06
10.14077/j.issn.1007-7812.2017.05.008
2017-03-05;
2017-05-14
国家自然科学基金资助(No.21243007)
张家荣(1993-),女,硕士,从事含能材料合成研究。E-mail:sonia610@126.com
