哌啶分子轴向和赤道构象的量子计算
2017-10-18邱学军潘林峰
邱学军,潘林峰
(中南民族大学 电子信息工程学院, 武汉 430074)
哌啶分子轴向和赤道构象的量子计算
邱学军,潘林峰
(中南民族大学 电子信息工程学院, 武汉 430074)
利用Gaussian 03软件对哌啶分子轴向构象和赤道构象进行了量子计算,首先,采用Hartree-Fock (HF)方法, 在6-311++G(d,p)高精度基组水平上获得了两种构象分子基态的优化结构、红外光谱和拉曼光谱,这些数据为预测分子结构提供重要的依据;其次,在分子基态稳定构象的基础上,利用Time-Dependent Density Functional Theory(TD-DFT), 在HCTH/6-311++G(d,p)高精度基组水平上进一步获得了两种构象的紫外可见光谱,分子激发态能量以及激发态和离子态激发布居信息;最后,由激发态和离子态激发布居信息分析获得了分子激发态电离的相关性.这些结果既可以为实验上区分和标示哌啶分子构象异构体提供依据,也可以为实验上预测哌啶分子的动力学机制提供证据.
哌啶; 激发态; 光谱; 电离
AbstractThe quantum calculations of axial and equatorial conformer of piperidine are carried out by Gaussian 03 software. Firstly, using Hartree-Fock (HF) method with 6-311++G(d,p) level, the optimized ground state molecular structures,IR and Raman spectrums of two conformers are acquired, which provide important basis for predicting molecular structure. Secondly, based on the stable ground state structures, the UV-VIS spectrums, excited state energy of neutral piperidine, and the excitation configurations of neutral and cation piperidine for two conformers are further obtained by Time-Dependent Density Functional Theory (TD-DFT) with HCTH/6-311++G(d,p) high-precision basis set level. Finally, the ionization correlations between excited states and cation states are analyzed. These results not only helpful for distinguishing and detecting conformational isomers, but also benefit to predict dynamics mechanism in piperidine.
Keywordspiperidine; excite state; spectrum; ionization
分子构象在蛋白质等生物大分子中普遍存在,深刻地影响着人类的生命活动,如蛋白质的空间构象影响着蛋白质分子的一级结构及氨基酸的排列顺序.了解和研究分子构象的物理化学性质和激发态动力学机理,对避免由于分子构象的生物学功能不同而引发的构象疾病的治疗有重要的意义.人们对于分子构象的研究最早也来自于蛋白质分子[1-3],20世纪60年代初,人们认识蛋白质分子并非刚体而具有确定的运动性,其构象运动性和构象灵活性对蛋白质功能的发挥具有重要意义.由于蛋白质分子结构的复杂性,目前无论从实验或者理论上来讲,对蛋白质构象动力学的实时观测依然具有相当大的困难.近年来,人们逐渐将研究重点转移到一些构象小分子体系上,21 世纪初,美国T A Miller小组在研究小分子构象动力学方面做了杰出的工作,他们利用激光诱导荧光光谱方法和近红外的腔衰荡光谱的方法对烷氧基的不同构象和丁基-过氧乙酸自由基的四个构象异构体进行了区分和标定[4-5].此外,美国的T S Zwier等利用受激发射泵浦-孔填充光谱和受激发射泵浦-诱导布居转移光谱的方法测得了色胺分子构象相互转化的阈值能量[6],2003年,美国的P M Weber教授首次提出用分子里德堡态能量的变化来探测分子结构的变化的方法[7], 并成功地对邻、间、对氟苯酚的分子结构进行了甄别[8].我国苏州大学的李宝宗教授采用HF/6-31G*方法对氨基型和亚胺型可乐定中性分子进行了构象分析[9].
哌啶分子是重要的饱和胺分子,在药物合成和DNA测序中有着广泛的应用.不仅如此,哌啶分子还是重要的构象分子,哌啶分子中的N-H键通过单键自由旋转可以形成两种不同的空间结构.即轴向构象和赤道式构象,二者在常温下的比例约为1∶3.近年来,关于哌啶分子的研究还主要集中在共振增强多光子电离光谱[10],吸收光谱和荧光光谱[11]等方面,而对于两种分子构象的理论,以及它们可能的激发态动力学过程的研究,目前仍然尚未开展.本文采用Gaussian 03[12]软件对哌啶分子轴向构象和赤道构象进行理论计算,得到了分子基态的稳定构型、红外光谱及拉曼光谱.此外,计算还获得了两种构象的紫外可见吸收光谱,分子激发态能量以及激发态和离子态激发布居信息.由激发态和离子态激发布居信息获得的分子激发态电离的相关性,可以为实验上光电子能谱的标定、预测分子的激发态的动力学机制提供重要的佐证.
1 理论计算方法
本文理论计算均在Gaussian 03中进行.首先采用HF方法, 在6-311++G(d,p)高精度基组水平上,得到了哌啶轴向和赤道两种构象分子的稳定结构、红外光谱和拉曼光谱.随后,利用TD-DFT方法, 在HCTH/6-311++G(d,p)高精度基组水平上进行能量计算,得到了这两种构象的紫外可见吸收光谱.此外,计算还获得了两种构象分子的激发态能量以及中性分子激发态和离子激发态的轨道跃迁信息,并由此得到了分子激发态与离子态的电离相关性.
2 理论计算结果
2.1哌啶分子轴向和赤道构象的基态结构
利用HF方法6-311++G(d,p)基组计算了哌啶分子两种构象的基态稳定结构,如图1所示,两种分子都具有Cs对称性,其中轴向构象分子的N—H键与哌啶分子环垂直,赤道构象分子的N—H键则与哌啶分子环平行,两种分子优化后的结构参数如表1所示.由表1可以看出,两种构象分子的键长是几乎相等的,然而一些特殊的键角和二面角是显著不同的,如对分子键角而言,轴向构象分子的A(7,10,16)和A(4,13,16)键角相对于赤道构象分子明显增大,而A(14,13,16) 和A(12,10,16) 键角则相对减小.对二面角而言,D(1,4,13,16),D(5,4,13,16),D(6,4,13,16),D(1,7,10,16),D(8,7,10,16)等均有较大的变化.观察这些键角和二面角发现,两种构象参数的变化都与N原子有关,因此主要是由于两种构象分子中N—H键的不同取向而引起的.
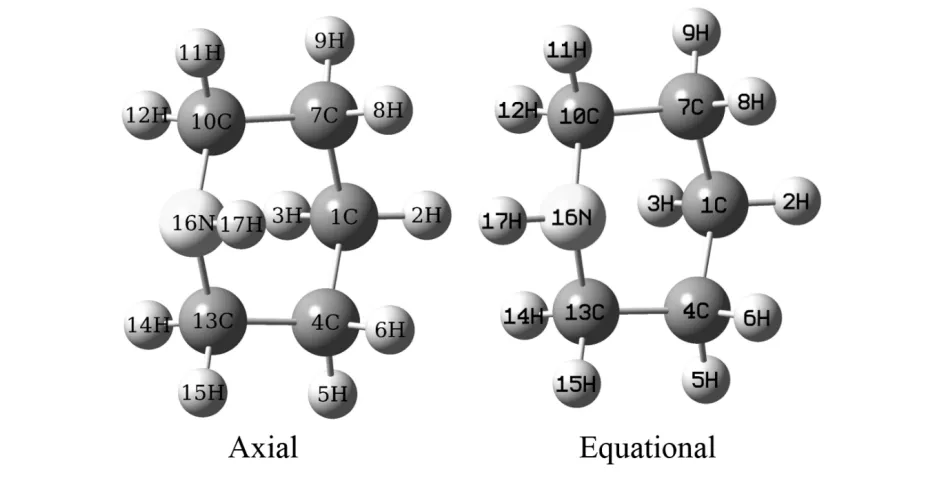
图1 优化得到的哌啶分子的基态结构Fig.1 The optimized molecular structures of piperidine in the ground state
2.2哌啶分子的轴向和赤道构象的红外光谱
图2所示为优化基态构象后得到的哌啶分子轴向和赤道构象的红外光谱,其中黑色曲线为理论计算结果,红色曲线是美国国家标准局(NIST)数据库提供的数据[13],由图可见,两种构象的理论红外光谱与实验结果基本吻合得很好,说明我们使用的计算方法和优化的分子基态结构是可信的.尽管如此,理论计算结果相对实验结果有整体红移,该红移可能和实验中所用的溶剂有关.从计算结果来看,轴向构象分子共有6个较强的振动特征峰,而赤道构象哌啶分子共有10个较强的特征峰,新出现的4个特征峰对应的波数分别为584.16 cm-1, 1130.99 cm-1, 1489.26 cm-1和3066.14 cm-1.这些振动对应的振动方式如图3所示,图中原子键轴处的箭头表示跃迁偶极矩的单位矢量,各原子处的箭头表示对应原子的位移矢量,其中584.16 cm-1, 1130.99 cm-1, 1489.26 cm-1对应N—H键和环上C—H键的摇摆振动,3066.14 cm-1则对应12H和14H原子的伸缩振动.
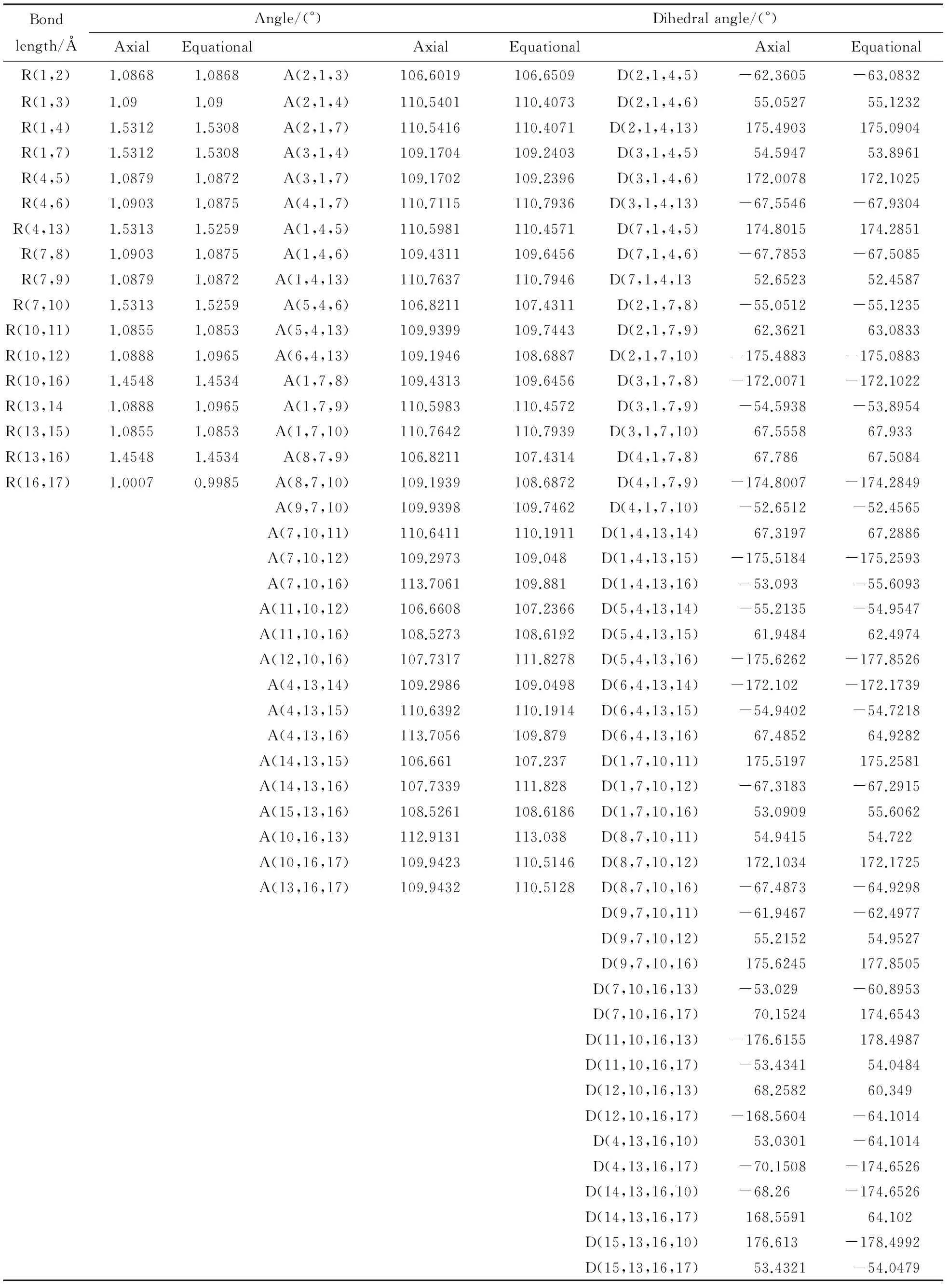
表1 哌啶分子两种构象的S0态优化几何参数对比Tab.1 Comparison between the optimized geometrical parameters of two conformers of piperidine in S0 state
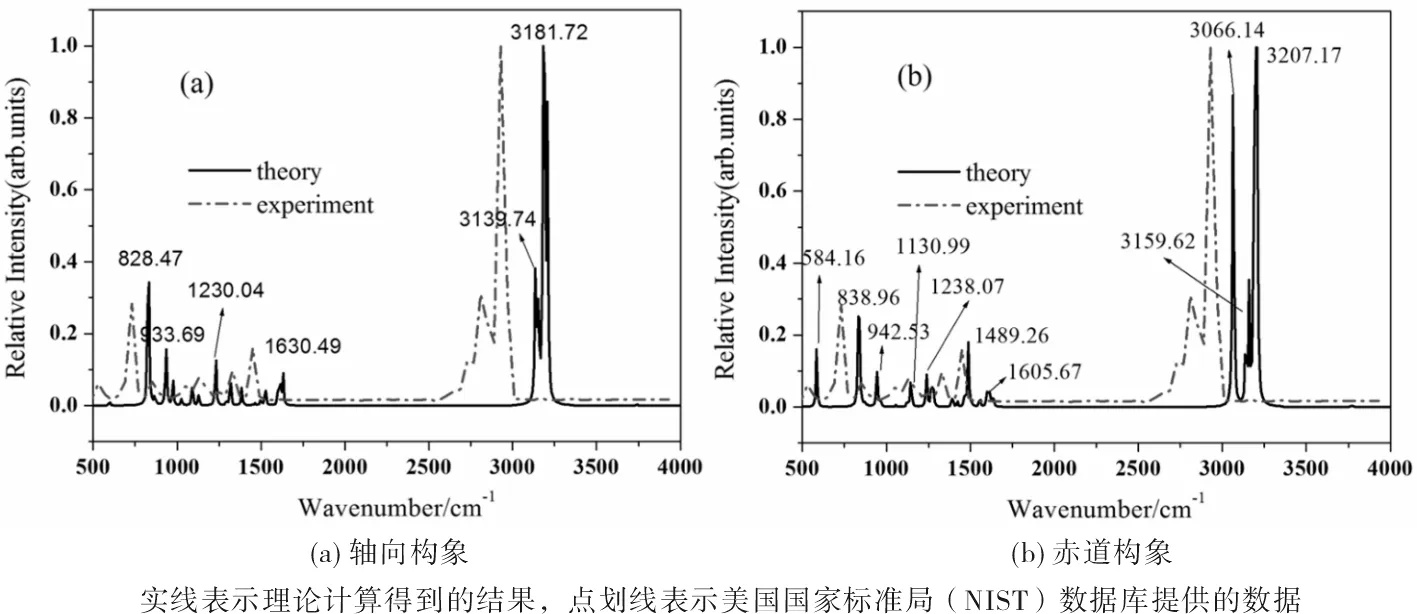
图2 哌啶分子的红外光谱Fig.2 The IR spectrum of piperidine

图3 哌啶赤道构象分子中额外的振动模式和频率Fig.3 The additional vibrational modes and frequency in equatorial conformer
2.3哌啶分子的轴向和赤道构象的拉曼光谱
计算同时获得了两种构象分子的拉曼光谱,如图4所示,计算结果显示, 哌啶轴向构象分子的拉曼活性主要分布在两个区域:一个位于低能区(750~1800 cm-1), 其中864 cm-1和 1600 cm-1对应于N—H键以及C—H的键的摇摆振动; 另一个在高能区(3000~4000 cm- 1), 3136 cm- 1,3152 cm- 1, 3184 cm- 1, 3208 cm- 1以及3744 cm- 1均对应于哌啶环上C—H键的伸缩振动.对于赤道构象的哌啶分子,低能区的拉曼活性基本与轴向构象分子相同,而高能区的强度与谱峰位置有一定的差异,这些差异主要是由于两种构象分子N—H键取向不同,导致原子在振动过程中极化率的变化不同而引起的.尽管如此,赤道构象分子各谱峰对应的振动模式基本和轴向构象分子相同,其中872 cm-1和 1600 cm-1对应于N—H键以及C—H的键的摇摆振动,3064 cm- 1, 3160cm- 1, 3200 cm- 1和3768 cm- 1则对应于哌啶环上C—H键的伸缩振动.

图4 哌啶分子的拉曼光谱 Fig.4 The Raman spectrum of piperidine
2.4哌啶分子的轴向和赤道构象的紫外吸收光谱
采用TD-DFT方法,基组为HCTH 6-311++G(d,p),对优化后的两种分子基态构象进行能量计算,如图5所示,得到了前20个激发态的紫外可见吸收光谱.由图可见,轴向构象分子主要有两个较强的特征吸收峰,分别对应的波长为166.2 nm和260.7 nm,其中最强峰对应的波长为166.2 nm,说明轴向构象分子对这一波长的光吸收最强.赤道构象分子两个主要吸收峰分别位于172.5 nm和248.1 nm处,对应的波长与轴向构象分子两个吸收峰相似.赤道构象在207.5 nm处还观察到了一个较强的吸收峰,这说明了哌啶两种构象分子在200 nm激光作用下,只有赤道构象分子能吸收光子而被激发.这些吸收谱特征为观察赤道构象分子激发态能量转移过程提供了可能.此外,不难发现,两种构象分子的吸收全部位于紫外区内,在可见光区均无吸收.Arthur M. Halpern小组曾利用实验获得了气相哌啶分子的吸收光谱,其实验结果与我们的理论结果可以很好的吻合.根据他们对特征吸收峰的标示[11],可以把248.1nm和207.5 nm处的特征吸收峰分别归属为S1←S0和S2←S0态的电子跃迁.由于轴向构象在S2态附近无吸收,在S1态附近的吸收强度也比赤道构象要弱得多,可以推断常温下哌啶分子在200 nm激光作用下的激发态的动力学过程可能主要由赤道构象主导.
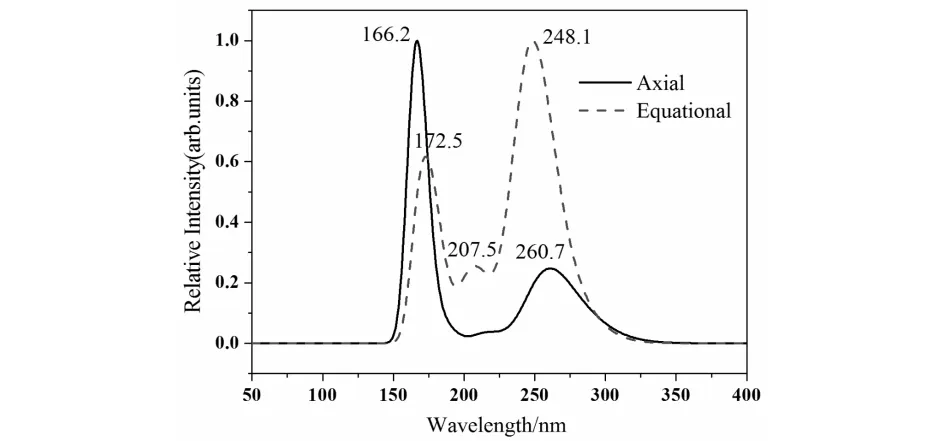
图5 哌啶分子两种构象的紫外-可见光谱Fig.5 The UV-VIS spectrum of piperidine
2.5哌啶分子的轴向和赤道构象的激发态与离子态的电离相关性
利用TD-DFT/HCTH 6-311++G(d,p)方法,获得了两种构象中性分子的激发态和离子态分子轨道,如图6所示, 对于两种构象中性分子的最高占据分子轨道(HOMO)电子,电子云主要分布在N原子周围,当分子被激发至最低未占分子轨道(LUMO)时,电子则较均匀地分布在N原子,H原子,C原子周围.而对于离子态的电子,两种构象分子的HOMO轨道电子云几乎有相同的分布,这暗示了这两种构象离子基态可能具有相同的能量.接下来,我们把两种构象和分子的中性激发态和离子激发态布居,跃迁系数,权重以及激发态能量总结在表2中,由表2可以看到,在两种构象的弗兰克-康登区域,S1态和S2态100%来自于HOMO-LUMO轨道的电子跃迁,与其它跃迁轨道无关,因此权重为1.0.由于只有离子基态为一个单个的HOMO轨道,根据库珀曼近似(Koopmans approximation), 可以假定分子在吸收紫外光后的电离过程与从HOMO轨道移去一个电子有关,对于两种构象分子的S1态和 S2态的电离而言,从LUMO轨道移去一个电子,将产生单个的HOMO轨道,刚好对应哌啶分子阳离子的基态,因此,在弗兰克-康登构型处应用库珀曼近似,可以将S1和S2态在不同激发能下的电离过程唯一的与D0关联起来.
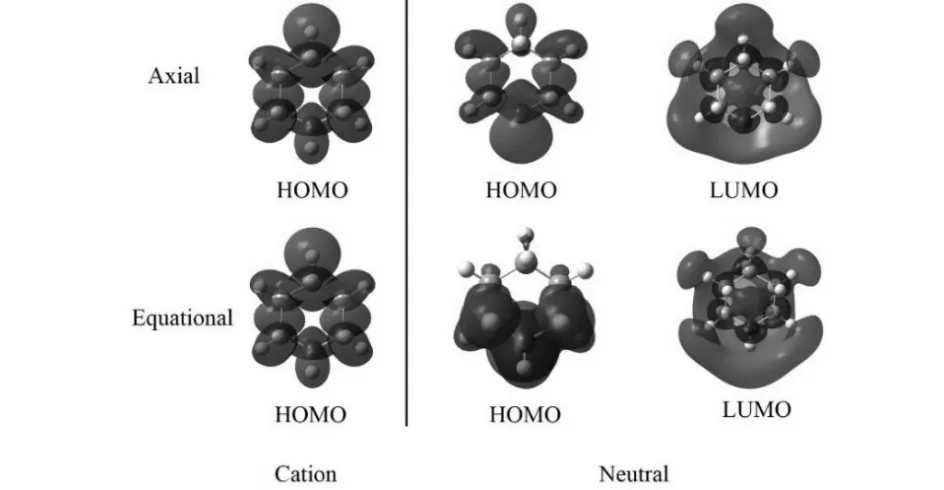
图6 哌啶分子两种构象的中性和离子态轨道Fig.6 Orbitals of neutral piperidine and the piperidine cation for two conformers
依据上述理论计算得到的结果,结合我们之前报道的光电子能谱数据[17],可以对哌啶分子的电离过程作如下分析,哌啶分子吸收两个400nm光子激发至S1态和S2态,由于分子的绝热电离势大概为7.85 eV[14], 处于激发态的分子接着吸收两个800 nm的光子即可达到离子基态D0而电离,到达D1(10.76eV)则需要吸收三个光子, 根据计算获得的分子轨道信息,这个概率是非常低的,因此,可以忽略来自离子激发态的电离.预计最大的光电子动能为1.45 eV.哌啶分子S1态和S2态的带源分别使用实验值4.76 eV和5.46 eV[11],根据弗兰克-康登原理,预计获得的光电子能谱峰的位置分别为0.01 eV和0.61 eV,这与实验结果吻合的非常好[17].此外,根据计算获得的轴向和赤道构象分子S1态和S2态的能量差别,分别在0.1 eV和0.3 eV左右,它们对应的光电子能谱峰位置会有较小的移动,然而在实验获得的光电子能谱中,除了1峰和2峰外,并未观察到其它明显的光电子能谱峰[17],这可能是由于轴向构象分子相对赤道构象分子在紫外区域的吸收要弱得多,在吸收飞秒激光后的动力学过程主要由赤道构象分子主导,其光电子能谱信号淹没在赤道构象分子的光电子能谱信号之中.综上所述,我们可以把光电子能谱所显示的1峰和2峰归属为赤道构象分子D0←S1和D0←S2的电离过程.由此可以预测哌啶分子在紫外光作用下的动力学机制是从S1高振动态向S2低振动态的内转换过程.这种动力学机制可能依赖于赤道型分子中较多的振动模式的激活,尤其是N原子和H原子对应振动模式的激活,可能导致S2高振动态的粒子向S2低振动态转移.
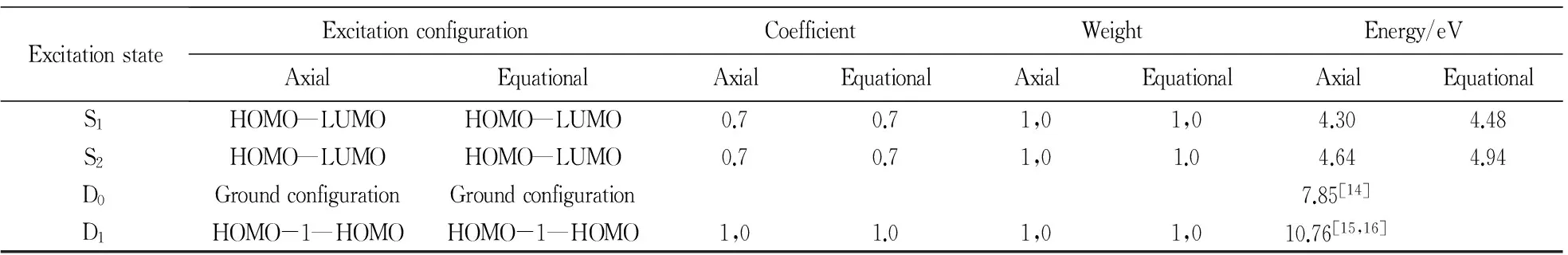
表2 哌啶轴向和赤道构象分子的S1,S2和D0态的能量和激发态布居Tab.2 Energies and configurations of S1,S2 and D0 states for axial and equatorial conformer piperidine
3 结语
利用Gaussian 03软件对哌啶分子的两种构象异构体(轴向构象和赤道构象)进行量子计算.首先采用HF方法, 在6-311++G(d,p)基组水平上获得了两种构象分子基态的稳定结构、红外光谱和拉曼光谱.其次,在分子基态稳定结构的基础上,利用TD-DFT算法, 在HCTH/6-311++G(d,p)基组水平上进一步获得了两种构象的紫外可见光谱,分子激发态能量以及激发态和离子态激发布居信息.最后由激发态和离子态激发布居信息分析了分子激发态电离的相关性及在紫外光作用下分子的动力学机制.
[1] Edsall J T,Flory P J,Kendrew J C,Liquori A M,Némethy G,Ramachandran G N,H.Seheraga A.A proposal of standard conventions and nomenclature for the description of polypeptide conformation [J].J Biol Chem,1966,241(4): 1004-1008.
[2] Edsall J T,Flory P J,Kendrew J C,Liquori A M,Némethy G,Ramachandran G N,Seheraga H A.A proposal of standard conventions and nomenclature for the description of polypeptide conformations [J].Biopolymers,1966,4(1): 121-130.
[3] Edsall J T,Flory P J,Kendrew J C,Liquori A M,Némethy G,Ramachandran G N,Scheraga H A.A proposal of standard conventions and nomenclature for the description of polypeptide conformations [J].J Mol Biol,1966,15(1): 399-407.
[4] Zu L,Liu J J,Gopalakrishnan S,Miller T A,The rotationally resolved electronic spectra of several conformers of 1-hexoxy and 1-heptoxy [J].Can J Chem,2004,82(6): 854-866.
[5] Glover B G,Miller T A.Near-IR cavity ring down spectroscopy and kinetics of the isomers and conformers of the butyl peroxy radical [J].J Phys Chem A,2005,109(49): 11191-11197.
[6] Dian B C,Clarkson J R,Zwier T S.Direct measurement of energy thresholds to conformational isomerization in tryptamine [J].Science,2004,303(5661): 1169-1173.
[7] Kuthirummal N,Weber P M.Rydberg states: sensitive probes of molecular structure [J].Chem Phys Lett,2003,378(5-6): 647-653.
[8] Gosselin J L,Weber P M.Rydberg fingerprint spectroscopy:a new spectroscopic tool with local and global structural sensitivity [J].J Phys Chem A,2005,109(22): 4899-4904.
[9] LI B Z.Theoretical investigation on conformational isomeri- zation and tautomerization of clonidine [J].Acta Chim Sinica,2006,64(4): 278-282.
[10] Streibel T,Hafner K,Muhlberger F,Adam T,Zimmermann R.Resonance-enhanced multiphoton ionization time-of-flight mass spectrometry for detection of nitrogen containing aliphatic and aromatic compounds: resonance-enhanced multiphoton ionization spectroscopic investigation and on-line analytical application [J].Appl Spectrosc,2006,60(1): 72-79.
[11] Halpern A M,Ramachandran B R,Glendening E D.The electronic spectroscopy and photophysics of piperidine in the vapor phase [J].J Phys Chem A,2000,104(50): 11733-11738.
[12] Frisch M J,Trucks G W,Schlegel H B,et al.Gaussian 03,Revision C.02 [CP].Wallingford CT: Gaussian,Inc,2004.
[13] National Institute of Standards and Technology[DB/OL].[2016-12-12].http://webbook.nist.gov/chemistry/
[14] Aue D H,Webb H M,Bowers M T.Quantitative proton affinities,ionization potentials,and hydrogen affinities of alkylamines [J].J Am Chem Soc,1976,98 (2): 311-317.
[15] Rozeboom M D,Houk K N.Stereospecific alkyl group effects on amine lone-pair ionization potentials: photoelectron spectra of alkylpiperidines [J].J Am Chem Soc,1982,104 (5): 1189-1191.
[16] Gan T H,Peel J B.Photoelectron spectroscopic studies of piperidine and its N-halo derivatives [J].Aust J Chem,1979,32 (3): 475-480.
[17] Qiu X J,Long J Y,Liu Z M,Zhang B.Direct imaging of the ultrafast internal conversion in isolated piperidine [J].Chem Phys Lett,2016,645: 133-137.
TheQuantumCalculationofAxialandEquatorialConformerofPiperidine
QiuXuejun,PanLinfeng
(College of Electronics and Information Engineering, South-Central University for Nationalities, Wuhan 430074, China)
O561.3
A
1672-4321(2017)03-0068-06
2017-03-01
邱学军(1985-), 男, 讲师, 博士, 研究方向:原子分子光物理, E-mail:2014006@mail.scuec.edu.cn
国家自然科学基金资助项目(11404411)
