煤及油母中常见C—X(X=N,S)的解离能
2017-10-16李璐樊红军胡浩权
李璐,樊红军,胡浩权
(1中国科学院大连化学物理研究所分子反应动力学国家重点实验室,辽宁 大连 116023;2大连理工大学化工学院,精细化工国家重点实验室,辽宁 大连 116024)
煤及油母中常见C—X(X=N,S)的解离能
李璐1,樊红军1,胡浩权2
(1中国科学院大连化学物理研究所分子反应动力学国家重点实验室,辽宁 大连 116023;2大连理工大学化工学院,精细化工国家重点实验室,辽宁 大连 116024)
煤及油页岩中除了碳、氢和氧原子是主要的组成元素外,氮和硫等杂原子大都以C—X(X=N,S)的键合形式存在,在其结构及转化利用中同样发挥重要的作用。认识C—X的解离能,有助于建立氮、硫热解过程中的迁移模型,丰富对煤及油母中常见化学键性质的认识,对发展高效清洁的能源利用技术至关重要。利用双杂化密度泛函方法,系统研究了煤及油母中典型 C—X键的解离能(BDE)范围。研究结果表明,煤及油母中常见 C—N和 C—S 键的 BDE 值范围分别是 154.1~55.7 kcal·mol−1和 83.0~56.6 kcal·mol−1。在热解过程中,苯硫类自由基以及苯胺类自由基会在初期产生,其次才是巯基、胺类自由基等侧链取代自由基脱落。C—S键的整体BDE值范围比其他类型化学键更低,各类化学键最低BDE值的高低顺序符合O—H > C—H > C—C > C—N > C—S > C—O的规律,其中,只有当有PhO·生成时,C—S > C—O,否则C—O > C—S。
煤;油母;解离能;焓;热力学;计算化学
Abstract:The carbon,hydrogen,oxygen are the majority elements in the structure of coal and oil shale,while the nitrogen and sulfur atoms,mostly in the form of C—X (X=N,S),still play a nonnegligible role in the structure and application of coal and oil shale.The homolytic bond dissociation enthalpies (BDE) of C—X for model compounds that are representative of the functionalities present in coal and kerogen were computed by using a double-hybrid method mPW2PLYP.The studies will be helpful for people to construct the transfer model of N and S in pyrolysis,and enrich the knowledge of chemical reaction of typical bonds in coal and kerogen.The BDE for C—N and C—S cover a range from 154.1—55.7 kcal·mol−1and 83.0—56.6 kcal·mol−1,respectively.The results suggest that the thiophenyl radical or phenylamino radical are the most favorable intermediates in the early pyrolytic stage.With the increase of temperature,the loss of sulfydryl and amidogen groups become feasible,and the ring cleavage reaction of five-membered ring (pyrrole and thiophene) through homolysis of C—N and C—S bond can also occur.The phenyl radical is the most difficult to form,and the directly bond dissociation of C—N bond in pyridine with six-membered ring is extremely unfavorable in the pyrolysis of coal and kerogen.Comparedto other types of bonds,C—S bond has the lowest general BDE range.The order of the lowest BDE for various bonds is O—H > C—H > C—C > C—N > C—S > C—O.The bond dissociation of C—O to generate PhO·radical is easier than to generate PhS· radical through homolysis of C—S,otherwise the BDE of C—O is higher than C—S.
Key words:coal;kerogen;bond dissociation enthalpies;enthalpy;thermodynamics;computational chemistry
引 言
能源是人类活动的物质基础,人类社会的前进与发展离不开优质能源的开采使用和先进能源技术的开发利用。我国缺油、少气和煤炭相对丰富的能源结构决定了煤在我国能源消费结构中的主导地位。油页岩是重要的石油补充能源,近年来得到广泛关注[1-2]。低温热解是一种煤及油页岩常用的加工利用方式,但是由于煤及油页岩本身的成分特点和现有工艺技术的缺陷,目前的热解技术普遍存在油气收率低、重质组分含量高等技术难题,并且加工过程中由组分中的氮和硫转化生成的NOx、H2S和SO2等被普遍认为是导致酸雨的重要原因[3],N2O则能够对臭氧层产生破坏[4],这些都会对环境造成极大的破坏。对煤及油页岩的油母结构中常见C—X(X=N,S)化学键在热场中的解离行为的深入认识有助于建立它们在热解过程中的硫、氮迁移模型[5],对于发展高效清洁的能源利用技术具有重要的指导意义。
由于煤及油母初始结构的复杂性和可变性,选择能代表其特征官能团性质的模型化合物进行研究,有助于从分子水平理解它们的化学性质。Ling等研究了含氮原子的喹啉和异喹啉[6]以及含硫原子的噻吩[7]和苯硫酚[8]的热解反应机理,并指出分子内氢转移反应在模型化合物的热解过程中对产物的形成和分配起到了非常重要的作用,苯硫酚热解比噻吩热解更容易生成H2S。Dubnikova等[9]详细研究了邻位喹啉自由基和邻位异喹啉自由基之间的异构化过程以及它们各自后续的热解反应路径,结果发现两种自由基在热解反应初期会经历分子内异构化反应迅速达到平衡,使得两种自由基的最终热解产物及分布完全一致,并且计算得出的稳定产物分布与实验结果有良好的一致性。Liang等[10]利用ReaxFF分子动力学模拟研究了木质素热解和加氢热解中的硫迁移机理,指出单纯热解中硫原子会以杂环结构形式存在于热解产物中,而在有H2条件下的加氢热解中,H2会促使硫原子从含硫杂环形态转变成苯硫酚/醇形态,从而更易于转变成H2S,以达到脱除硫的目的。并且,在脱硫过程中,合理控制温度可达到最大限度脱硫的目的。
除了上述对含氮或硫模型化合物的具体产物生成机理研究之外,对于C—X(X=N,S)键解离能(bond dissociation enthalpies,BDE)的研究也引起了广泛关注。Khrapkovskii等[11]考察了多种方法水平下硝基苯和取代硝基苯类化合物的C—NO2键的BDE值,提出B3LYP/6-31G(d,p)方法的计算结果能很好地吻合实验值,苯环上的吸电子取代基会使C—NO2键的 BDE值降低。Yu等[12]评价了多种密度泛函方法(density functional theory,DFT)和基组在计算有机反应中常见的C—SO2R键解离BDE值时的表现,指出 M06-2X/6-31G(d)的计算结果最为可靠。Yao等[13]则通过对多种化学键型(C—H、O—H、C—C、C—N、C—S等)BDE值的计算,得出MPW1P86方法在多种DFT方法中表现最为可靠。由此可见,虽然已有文献关注过C—S和C—N键的反应性质[14-22],但研究所涉及到的煤及油母结构中的C—N和C—S键类型并不完善,并且未对其解离行为规律进行系统研究。此外,各文献选择的最优理论方法并不一致,有关煤及油母中化学键的 BDE也并未在同一计算水平下得到,以致不能直接采用已有数值进行相互之间的比较。鉴于热场中的键解离是反应的第一步,因此本文选取合理的模型化合物,对煤及油母中 C—X(X=N,S)的BDE值进行系统研究,其结果势必会对理解N和S在后续过程中的迁移途径有积极的推进作用,并能补充对整个煤及油母中各个类型化学键热反应性质的认识。
1 计算方法
分子A—B的键解离能(BDE)被定义为在标准状况下,气相反应式(1)中分子A—B键均裂生成A·和B·两个自由基所需的能量[23]

BDE的值只与反应物和生成物的相对焓值有关,可通过式(2)的计算获得
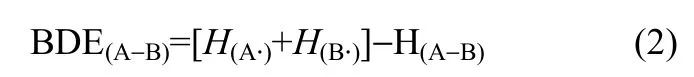
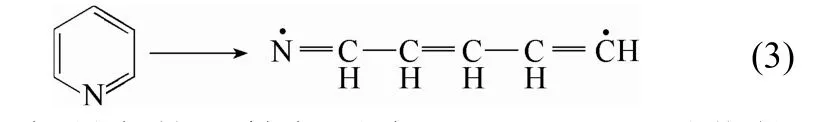
其中,H(A—B)为分子A—B的标准摩尔生成焓,H(A·)和H(B·)分别为自由基 A·和 B·的标准摩尔生成焓。吡啶等杂环开环的键解离能则通过键断裂后形成双自由基与母体分子的焓值差进行计算,以吡啶为例,其开环的C—N键的BDE值由反应式(3)的能量决定
本文中所有的计算都是在Gaussian 09量化软件包下完成,所有分子的优化都是在没有对称性限制下进行,每一个优化结构经过频率分析确认没有虚频。根据作者前期对解离能计算方法评估的研究结果[24],本文中选取双杂化密度泛函mPW2PLYP[25]方法在 cc-pVDZ[26]基组下进行构型优化和频率计算,并对优化结构在 cc-pVTZ[27]基组下进行了单点能量计算。所有的热力学校正数据均由频率计算的结果获得。为了使计算结果更准确,计算中包括了基组重叠误差(basis set superposition error,BSSE)校正[28-29]。本文中所有能量单位均为 kcal·mol−1(1 kcal·mol−1=4.184 kJ·mol−1)。
文中选择了煤及油母中常见C—N和C—S键的模型化合物进行研究,所选择的化合物结构见图1。主要包括含氮或含硫侧链以及桥键的芳烃化合物、含氮或含硫杂环化合物,如吡啶、吡咯和噻吩等。

图1 所选模型化合物Fig.1 Model compounds under study in this work
2 实验结果与讨论
2.1 C—N键的解离能范围
表1为计算得到的不同化合物中 C—N键的BDE值。C—N键的 BDE值范围为 154.1~55.7 kcal·mol−1。其中,吡啶环的C—N键断裂的解离能最高,达到 154.1 kcal·mol−1,而 PhCH2—NHPh 键断裂的BDE值最低。与先前研究报道的C—C键型和C—O键型的BDE值规律相似[30],生成Ph·自由基的 C—N键解离能量较高,其 BDE值范围为82.1~99.8 kcal·mol−1。有 PhCH2·自由基和类苄基的o,m,p-C5H4NCH2·自由基生成的 C—N键的断裂所需能量最低,其 BDE值范围为 55.7~71.2 kcal·mol−1。化合物中甲基取代的胺类自由基解离出CH3·自由基的 C—N 键 BDE值范围为 63.2~73.3 kcal·mol−1,其中处于桥键位置的 N—CH3键解离比苯环侧链取代的N—CH3键解离所需能量更低,因此更易发生。由吡啶环替代苄胺中苯环得到的o,m,p-C5H4NCH2—NH2中的C—N键的BDE值分别比PhCH2—NH2高 1.8、0.6 和 1.1 kcal·mol−1。值得注意的是,不同于吡啶六元环开环需要非常高的能量值,吡咯的 C—N键解离所需能量为 84.5 kcal·mol−1。各种 C—N 键的 BDE 值的顺序见图2。

表1 不同化合物中C—N键的解离能Table 1 BDE of C—N bonds/kcal·mol−1
2.2 C—S键的解离能范围
表2中列出了计算得到的不同化合物中 C—S键 BDE值。C—S键的BDE值范围为 83.0~56.6 kcal·mol−1。其中,Ph—SH 键断裂产生的 BDE 值最高,而PhCH2—SH键断裂产生的BDE值最低。与C—N键型的BDE值规律相似,生成Ph·自由基的C—S键解离能量较高,其BDE值范围为80.0~83.0 kcal·mol−1。有 PhCH2·自由基生成的 PhCH2—S 键的断裂所需能量最低,其 BDE值范围为 56.6~67.4 kcal·mol−1。显而易见,这些 C—S 键的 BDE 值明显低于相应的C—N键BDE值。噻吩的C—S键断裂所需能量为 80.8 kcal·mol−1,该值与吡咯的 C—N 键BDE值较为接近。邻位取代的吡啶环上发生甲硫基解离所需能量明显低于间位和对位取代,该现象与Hayes等[31]计算的甲基在吡啶环上不同取代位的解离能规律一致。各种C—S键的BDE值的顺序见图3。

图 2 C—N 键的解离能区间(50~155 kcal·mol−1)Fig.2 C—N BDE range from 50 to 155 kcal·mol−1
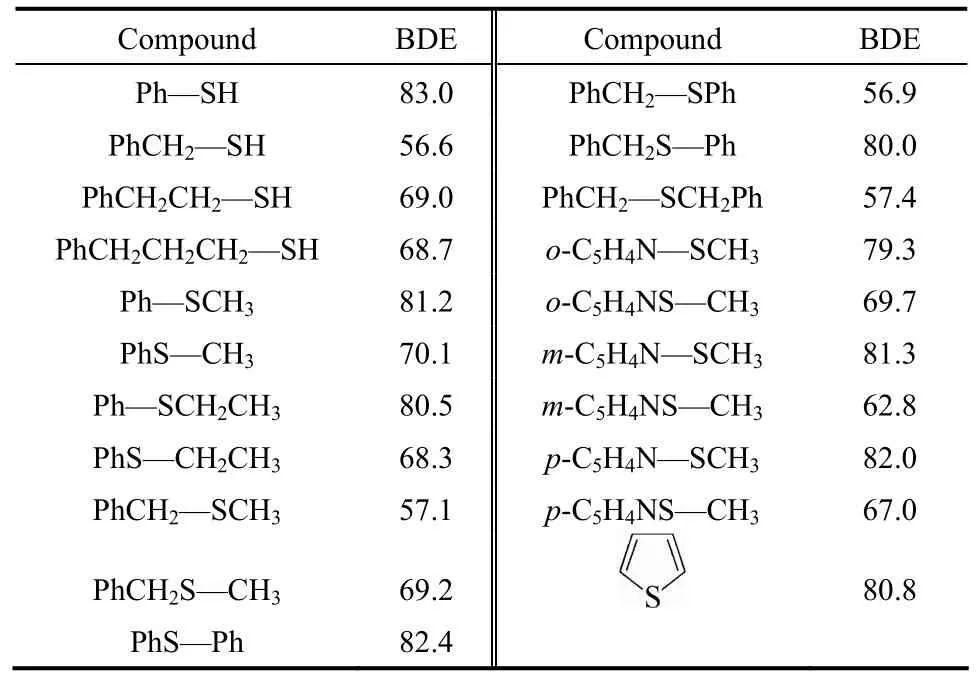
表2 不同化合物中C—S键的解离能Table 2 BDE of C—S bonds/kcal·mol−1
2.3 煤及油母中常见化学键的解离能范围
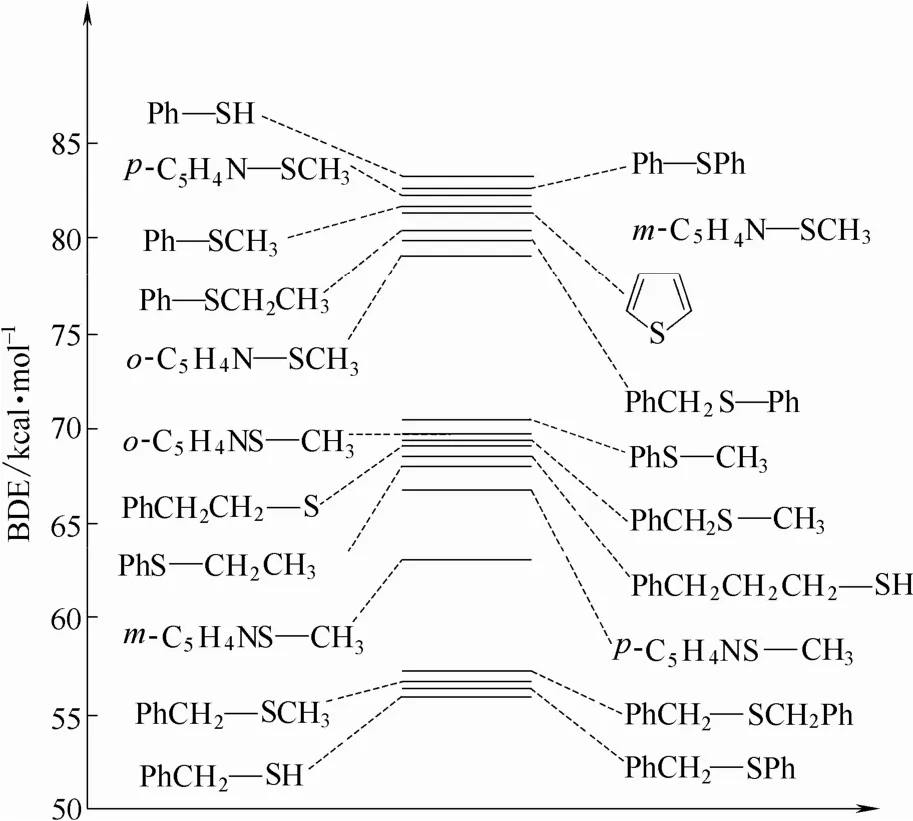
图 3 C—S 键的解离能区间(50~85 kcal·mol−1)Fig.3 C—S BDE range from 50 to 85 kcal·mol−1

图4 煤及油母中常见键的解离能区间(括号中的数值为所计算键型的数量)Fig.4 BDE range of different bond types in coal and kerogen (data in parentheses is number of bond type calculated)
结合先前的研究结果[30],可以得到煤及油母中几种常见化学键的BDE值范围(图4)。C—H、C—C、C—O、O—H、C—N和C—S键的BDE值范围分别为 111.4~81.2 kcal·mol−1、102.9~62.8 kcal·mol−1、107.6~52.6 kcal·mol−1、111.2~86.6 kcal·mol−1、154.1~55.7 kcal·mol−1和 83.0~56.6 kcal·mol−1。可以看出,每一种键型的 BDE值都是一个很大的范围,并且不同键型之间的 BDE值存在很大程度的重叠。有H·自由基生成的O—H和C—H键的BDE值普遍较高,C—C、C—O、C—N和 C—S键的最低 BDE值均来自于 PhCH2—YPh(Y=CH2、O、NH 和 S)。C—C、C—N 和 C—S键的高能量区域均由生成苯基和吡啶杂环的芳环自由基的键解离反应组成,而在C—O键的BDE值区间内,芳香羧酸类及芳香脂类化合物中羧基基团中C—O键断裂所需能量最高,其次才是有芳环自由基生成相关的C—O解离。从图4可以看出,每种键型的最低BDE值的高低顺序为O—H > C—H >C—C > C—N > C—S > C—O,结合先前研究成果,将同种类型化学键断裂的C—O和C—S解离能进行比较发现,有PhS·自由基生成的C—S键的BDE值总是比有PhO·生成的C—O键的解离能高,即C—SPh > C—OPh,这得益于PhO·自由基中的O可以与苯环形成良好的共轭,使得PhO·自由基具有更高的稳定性,从而有更低的 BDE值。而在其他同种类型的BDE值比较中,C—S键的BDE值均低于对应的C—O键的BDE值。Shi等[32]提出脂肪醚和脂肪醇类化合物中的C—O键键长远小于脂肪硫醚和硫醇类化合物中的C—S键键长是导致这一现象的直接原因。
2.4 热解中化学键的断裂顺序
键解离是热解过程的第一步,解离能的大小直接关系煤及油母中化学键断裂的先后顺序,从而影响后续的整个热解过程,因此对煤及油母中常见C—X(X=N,S)化学键解离能的系统研究不仅有利于了解N,S原子的迁移途径,而且对煤及油母结构中化学键以及热解反应过程的认识具有重要的作用。
综合先前对煤中常见4种类型(C—H、C—C、C—O和O—H)化学键BDE值的研究结果[30]可知,煤及油母热解过程中应首先断裂由苯氧类自由基、苄基类自由基、苯硫类自由基以及苯胺类自由基相互连接而形成的弱桥键。随着温度的逐渐升高,芳环的侧链取代基开始解离,巯基相比其他小自由基更易解离,随后是胺类自由基、烷氧类自由基和烷基类自由基的生成,而氢自由基的直接生成较为困难。同时,在此温度区间内,五元环的吡咯和噻吩也有可能会发生开环反应。此外,生成苯自由基或类似的芳环自由基同样需要较高的能量,因此这类反应只能在高温下发生。由于六元环的吡啶直接开环所需的键解离能量过高,可以推测该六元芳香环并不是通过直接键断裂的开环反应参与热解反应过程。
3 结 论
(1)煤及油母中常见C—N和C—S键的BDE值范围分别是 154.1~55.7 kcal·mol−1和 83.0~56.6 kcal·mol−1,与 C—H、C—C、C—O 和 O—H 键的BDE值范围间同样存在很大程度的重叠。
(2)煤及油母中的N和S倾向于首先形成苯硫类自由基以及苯胺类自由基,巯基比胺类自由基更易形成。
(3)煤及油母中常见键型的最低 BDE值的高低顺序符合O—H > C—H > C—C > C—N > C—S >C—O的规律。当有PhO·生成时,C—S > C—O,否则 C—O > C—S。
[1]柏静儒,林卫生,潘朔,等.油页岩低温热解过程中轻质气体的析出特性[J].化工学报,2015,66(3): 1104-1110.BAI J R,LIN W S,PAN S,et al.Characteristics of light gases evolution during oil shale pyrolysis[J].CIESC Journal,2015,66(3):1104-1110.
[2]DYNI J R.Geology and resources of some world oil shale oil deposits[J].Shale,2003,20(3): 193-252.
[3]GAFFNEY J S,STREIT G E,SPALL W D,et al.Beyond acid rain.Do soluble oxidants and organic toxins interact with SO2and NOxto increase ecosystem effects [J].Environmental Science Technology,1987,21(6): 519-524.
[4]余岳溪,高正阳,季鹏,等.煤焦异相还原N2O的反应机理[J].化工学报,2017,68(1): 369-374.YU Y X,GAO Z Y,JI P,et al.Heterogeneous reduction reaction of N2O by char[J].CIESC Journal,2017,68(1): 369-374.
[5]谢建军,杨学民,吕雪松,等.煤热解过程中硫氮及迁移规律研究进展[J].化工进展,2004,23(11): 1214-1218.XIE J J,YANG X M,LÜ X S,et al.Progress on transformation behavior of sulfur and nitrogen during coal pyrolysis[J].Chemical Industry and Engineering Progress,2004,23(11): 1214-1218.
[6]LING L,ZHANG R G,WANG B J,et al.Pyrolysis mechanisms of quinoline and isoquinoline with density functional theory[J].Chinese Journal of Chemical Engineering,2009,17(5): 805-813.
[7]LING L,ZHANG R G,WANG B J,et al.Density functional theory study on the pyrolysis mechanism of thiophene in coal[J].Journal of Molecular Structure (THEOCHEM),2009,905(1/2/3): 8-12.
[8]LING L,ZHANG R G,WANG B J,et al.DFT study on the sulfur migration during benzenethiol pyrolysis in coal[J].Journal of Molecular Structure (THEOCHEM),2009,952(1/2/3): 31-35.
[9]DUBNIKOVA F,TAMBURU C,LIFSHITZ A.A deep insight into the details of the interisomerization and decomposition mechanism ofo-quinolyl ando-isoquinolyl radicals.Quantum chemical calculations and computer modeling[J].The Journal of Physical Chemistry A,2016,120(38): 7538-7547.
[10]LI G Y,WANG F,WANG J P,et al.ReaxFF and DFT study on the sulfur transformation mechanism during the oxidation process of lignite[J].Fuel,2016,181: 238-247.
[11]KHRAPKOVSKII G M,SHARIPOV D D,SHAMOV A G,et al.Theoretical study of substituents effect on C—NO2bond strength in mono substituted nitrobenzenes[J].Computational and Theoretical Chemistry,2013,1017: 7-13.
[12]YU H Z,FU F,ZHANG L,et al.Accurate predictions of C—SO2R bond dissociation enthalpies using density functional theorymethods[J].Physical Chemistry Chemical Physics,2014,16:20964-20970.
[13]YAO X Q,HOU X J,JIAO H J,et al.Accurate calculations of bond dissociation enthalpies with density functional methods[J].The Journal of Physical Chemistry A,2003,107(46): 9991-9996.
[14]DEL GIACCO T,LANZALUNGA O,LAPI A,et al.Photosensitized oxidation of aryl benzyl sulfoxides.Evidence for nucleophilic assistance to the C—S bond cleavage of aryl benzyl sulfoxide radical cations[J].Journal of Organic Chemistry,2015,80(4): 2310-2318.
[15]KHRAPKOVSKII G M,SHARIPOV D D,SHAMOV A G,et al.Enthalpies of formation of mono substituted nitrobenzenes: a quantum chemistry study[J].Computational and Theoretical Chemistry,2013,1011: 37-43.
[16]FAYET G,ROUTUREAU P,MINISINI B.Decomposition mechanisms of trinitroalkyl compounds: a theoretical study from aliphatic to aromatic nitro compounds[J].Physical Chemistry Chemical Physics,2014,16(14): 6614-6622.
[17]MAROCHKIN I I,DOROFEEVA O V.Amide bond dissociation enthalpies: effect of substitution on N—C bond strength[J].Computational and Theoretical Chemistry,2012,991: 182-191.
[18]JOHNSON E R,CLARKIN O J,DILABIO G A.Density functional theory based model calculations for accurate bond dissociation enthalpies(3): A single approach for X—H,X—X,and X—Y (X,Y=C,N,O,S,halogen) bonds[J].The Journal of Physical Chemistry A,2003,107(46): 9953-9963.
[19]SONG X L,PARISH C A.Pyrolysis mechanisms of thiophene and methylthiophene in asphaltenes[J].The Journal of Physical Chemistry A,2011,115(13): 2882-2891.
[20]ZHENG W R,FU Y,GUO Q X.G3//BMK and its application to calculation of bond dissociation enthalpies[J].Journal of Chemical Theory and Computation,2008,4(8): 1324-1331.
[21]TAO X X,TANG L F,XIE M H,et al.Dielectric properties analysis of sulfur-containing models in coal and energy evaluation of their sulfur-containing bond dissociation in microwave field[J].Fuel,2016,181: 1027-1033.
[22]TANG L F,WANG S W,GUO J F,et al.Exploration on the removal mechanism of sulfur ether model compounds for coal by microwave irradiation with peroxyacetic acid[J].Fuel Processing Technology,2017,159: 442-447.
[23]BARCKHOLTZ C,BARCKHOLTZ T A,HADAD C M.C–H and N—H bond dissociation energies of small aromatic hydrocarbons[J].Journal of the American Chemical Society,1999,121(3): 491-500.
[24]LI L,FAN H J,HU H Q.Assessment of contemporary theoretical methods for bond dissociation enthalpies[J].Chinese Journal of Chemical Physics,2016,29(4): 453-461.
[25]SCHWABE T,GRIMME S.Towards chemical accuracy for the thermodynamics of large molecules: new hybrid density functionals including non-local correlation effects[J].Physical Chemistry Chemical Physics,2006,8(38): 4398-4401.
[26]DUNNING J T H.Gaussian basis sets for use in correlated molecular calculations(Ⅰ): The atoms boron through neon and hydrogen[J].The Journal of Chemical Physics,1989,90(2): 1007-1023.
[27]KENDALL R A,DUNNING J T H,HARRISON R J.Electron affinities of the first-row atoms revisited.Systematic basis sets and wave functions[J].The Journal of Chemical Physics,1992,96(9):6796-6806.
[28]BOYS S F,BERNARDI F.Calculation of small molecular interactions by differences of separate total energies—some procedures with reduced errors[J].Molecular Physics,1970,19(4):553.
[29]SIMON S,DURAN M,DANNENBERG J J.How does basis set superposition error change the potential surfaces for hydrogen bonded dimers[J].Journal of Chemical Physics,1996,105(24): 11024-11031.
[30]LI L,FAN H J,HU H Q.A theoretical study on bond dissociation enthalpies of coal based model compounds[J].Fuel,2015,153: 70-77.
[31]HAYES C J,HADAD C M.Combustion pathways of the alkylated heteroaromatics: bond dissociation enthalpies and alkyl group fragmentations[J].The Journal of Physical Chemistry A,2009,1133(45): 12370-12379.
[32]SHI J,HU X R,LIANG S.A computational study of C—S bond dissociation enthalpies in petroleum chemistry[J].Heteroatom Chemistry,2011,22(2): 97-105.
Bond dissociation enthalpies of C—X (X=N,S) in coal and kerogen
LI Lu1,FAN Hongjun1,HU Haoquan2
(1State Key Laboratory of Molecular Reaction Dynamics,Dalian Institute of Chemical Physics,Chinese Academy of Sciences,Dalian116023,Liaoning,China;2State Key Laboratory of Fine Chemicals,Institute of Coal Chemical Engineering,School of Chemical Engineering,Dalian University of Technology,Dalian116024,Liaoning,China)
TQ 021.2
A
0438—1157(2017)10—3900—06
10.11949/j.issn.0438-1157.20170377
2017-04-10收到初稿,2017-06-23收到修改稿。
联系人:樊红军,胡浩权。
李璐(1990—),女,博士后。
国家重点研发计划项目(2016YFB0600301)。
Received date:2017-04-10.
Corresponding author:Prof.FAN Hongjun,fanhj@dicp.ac.cn;Prof.HU Haoquan,hhu@dlut.edu.cn
Foundation item:supported by the National Key R&D Program of China (2016YFB0600301).
