NHPI催化分子氧选择性氧化木质素模型分子中的C—C键
2017-10-16刘长军陈宁刘颖颖鲁厚芳梁斌唐思扬岳海荣谭平华
刘长军,陈宁,刘颖颖,鲁厚芳,梁斌,,唐思扬,岳海荣,谭平华
(1四川大学化学工程学院,四川 成都 610207;2四川大学CCUS与CO2矿化利用研究中心,四川 成都 610207;3西南化工研究设计院有限公司,四川 成都 610225)
NHPI催化分子氧选择性氧化木质素模型分子中的C—C键
刘长军1,陈宁1,刘颖颖2,鲁厚芳1,梁斌1,2,唐思扬1,岳海荣1,谭平华3
(1四川大学化学工程学院,四川 成都 610207;2四川大学CCUS与CO2矿化利用研究中心,四川 成都 610207;3西南化工研究设计院有限公司,四川 成都 610225)
以1,2-二苯乙烷为模型分子,考察了N-羟基邻苯二甲酰亚胺(NHPI)对分子氧氧化断裂木质素分子中C—C键的催化作用,比较了Cu、Co和Mn的多种可溶性盐对NHPI催化活性的促进作用,探究了工艺条件对1,2-二苯乙烷氧化的影响规律。结果表明:NHPI能够在温和条件下催化分子氧氧化1,2-二苯乙烷中的C—C键断裂生成苯甲醛和苯甲酸。可溶性的Cu、Co和Mn盐都具有助催化作用,其中乙酸锰的助催化作用最强。确定了二苯基乙酮为氧化的初级反应产物。发现提高反应温度、增加乙酸锰用量以及延长反应时间,有利于C—C键的氧化断裂和提高小分子产物的选择性。优化的反应条件为NHPI为底物物质的量的10%,乙酸锰和NHPI之比为1:10,反应温度90℃,氧气分压1.0 MPa,反应时间8 h,C—C键的氧化断裂产物苯甲醛和苯甲酸的总选择性可达76.5%。
生物质;催化;氧化;降解;生物燃料
Abstract:The catalytic effects ofN-hydroxyl phthalimide combined with various transition metal salts on the oxidative breakage of C—C bond of lignin by O2under mild conditions were studied.1,2-diphenylethane was adopted as the model compound of lignin C—C bond.It was found that Mn(OAc)2showed the highest promotion effect on the catalytic activity of NHPI among the soluble cobaltous,manganous and cupric salts.The oxidative breakage of C—C bonds led to the production of benzyl aldehyde and benzoic acid with high selectivity.1,2-Diphenyl ethanone was found to be the primary product.Either increase the reaction temperature,Mn(OAc)2amount as well as the reaction time favored the C—C bond cleavage.Total selectivity of benzyl aldehyde and benzoic acid up to 76.5% was achieved in acetic acid under the conditions of 90℃ and 1.0 MPa O2for 8 h by using 10% NHPI and 1% Mn(OAc)2with respect to the amount of benzyl phenyl ether as the catalyst.
Key words:biomass;catalysis;oxidation;degradation;biofuel
引 言
木质纤维素是储量最大、分布最广的生物质资源[1]。其中木质素约占木质纤维素总质量的 25%,是造纸制浆工业和纤维素乙醇工业的主要副产物。木质素结构稳定、难以转化,目前仍然难以进行高效的加工和利用,大多数木质素都被当作低值燃料烧掉[2]。探索木质素的高效转化途径实现高值利用对于提高木质纤维素资源利用率、提高制浆业和纤维素乙醇装置的经济性具有重要意义。木质素具有苯环结构且含氧量低[3],是制备高热值液体燃料和芳香族化学品的理想原料[4-6]。木质素是由愈创木基、紫丁香基和对羟基苯基3类苯丙烷结构单元,通过多种醚键(α-O-4、β-O-4和4-O-5等)和碳碳键(β-β、β-5和5-5等)交联共聚而成的天然高分子[7]。天然木质素中醚键占 60%~75%,碳碳键占20%~35%,在木质素提取过程会破坏大部分的芳醚键,使得提取出的木质素中醚键含量降低至10%左右,而碳碳键在该过程具有较高的稳定性[8-10]。
催化氧化能显著降低木质素主要化学键断裂的能垒,实现木质素高效转化,反应生成芳香醛、酸等高附加值的多官能团小分子有机化合物[6,11]。硝基苯[6]、金属氧化物[6]、氧气[11]、双氧水[3,12-13]和过氧乙酸[14]等都可以作为木质素氧化的氧化剂。Ouyang等[15]发现在甲醇水溶液中CuO/Fe2(SO4)3/NaOH能催化双氧水氧化木质素,并且单酚产物收率达到17.9 %。Yang等[16]发现在MgO催化下双氧水和氧气氧化木质素对紫丁香基产物和愈创木基产物具有不同的选择性,并且氧气比双氧水的活性更高。分子氧是原子经济性最高、廉价易得的理想氧化剂。高效的氧化催化剂是实现温和条件下分子氧氧化的关键[17]。Rahimi等[17]发现4-乙酰氨基-2,2,6,6-四甲基吡啶氮氧化物与硝酸和盐酸的稀溶液构成的催化体系对氧气氧化木质素具有较高活性。Hanson等[18-28]对多种钒配合物催化氧气氧化木质素解聚过程进行了系统的研究,发现钒配合物在吡啶中的催化活性最高,氧化反应主要导致碳氧键的断裂,而同样条件下氯化亚铜催化剂则主要是氧化碳氢键并导致碳碳键的断裂[27-28]。但这些钒催化剂主要是以进攻木质素中的醚键为主,对碳碳键的氧化解聚过程研究相对较少。钒配合物配体复杂,该催化剂不易获得并且须以吡啶为溶剂,因此该过程并非一个绿色的氧化过程。
N-羟基邻苯二甲酰亚胺(NHPI)中的O—H容易均裂形成邻苯二甲酰亚胺氮氧自由基(PINO),PINO能够夺取C—H键上的H形成烃基自由基,烃基自由基易与氧气结合形成烃基过氧化物,从而实现底物的分子氧氧化[29-30]。因此NHPI在烃类的液相氧化反应中表现出很好的催化活性[31-32]。向该体系中引入过渡金属盐能够促进PINO自由基的形成从而提高NHPI的催化活性[33]。Shiraishi等[34]发现 NHPI对木质素中α碳的电化学氧化羰基化也具有很好的催化作用。因此NHPI很可能对木质素特别是木质素中碳碳键氧化断裂也具有很好的催化活性。同时乙酸既是NHPI催化氧化过程的良溶剂也是提取木质素的良溶剂[32,35]。研究NHPI对木质素的氧化解聚的催化作用和反应规律,对开发新的木质素氧化体系以及探索木质素提取和氧化解聚过程耦合的理论基础具有重要意义。
本文以 1,2-二苯乙烷为木质素模型分子,研究以NHPI为主催化剂、可溶性过渡金属盐为助剂的复合催化剂对分子氧氧化断裂木质素分子中碳碳键的催化作用。采用气相色谱-质谱联用仪和气相色谱仪分别对液相产物进行了定性和定量分析。考察了过渡金属盐种类、催化剂用量、反应温度、氧气分压等因素对 1,2-二苯乙烷转化率和氧化产物选择性的影响,确定了优化的反应条件,可为分子氧催化氧化木质素的工艺开发提供必要的理论基础。
1 实验部分
1.1 仪器与试剂
福立9790Ⅱ型气相色谱分析仪,浙江福立分析仪器有限公司;100 ml Parr 4598HPHT型高压反应釜,美国 PARR公司;岛津气质联用仪(GCMS-QP2010 Plus),日本岛津公司。
1,2-二苯乙烷,质量分数≥97%,萨恩化学技术(上海)有限公司;N-羟基邻苯二甲酰亚胺,质量分数≥99%,成都市贝斯特试剂有限公司;邻硝基甲苯,质量分数≥99%,成都市艾科达化学试剂有限公司;二苯乙二酮,质量分数≥98.0%,上海腾准生物科技有限公司;四水乙酸锰,质量分数≥99.0%,天津市大茂化学试剂厂;苯甲醛(质量分数≥99.0%)、苯甲酸(质量分数≥99.5%)、冰醋酸、四水乙酸锰、四水乙酸钴、四水乙酸镍、一水乙酸铜、三水硝酸铜、无水硫酸铜和二水氯化铜,均为分析纯,购自于成都科龙化学试剂厂;氧气为工业级,体积分数>99%,购自于四川双流旭源气体有限公司。
1.2 实验方法
1,2-二苯乙烷氧化反应在 100 ml Parr 4598HPHT型高压反应釜中进行。称取 0.5 g(2.75×10−3mol)1,2-二苯乙烷和 30.0 g 冰酸酸加入反应釜中,再分别加入 0.0448 g(2.75×10−4mol)NHPI和 0.0067 g(2.75×10−5mol)四水乙酸锰。将高压反应釜密闭后,用0.5 MPa氧气置换釜内气体3次排出釜内空气后,再充入0.5 MPa的氧气。在40 min内将反应釜从室温升至 90℃后补充氧气至表压1.0 MPa,在反应过程中保持总压不变,搅拌转速为400 r·min−1,反应时间10 h。反应结束后,自然冷却至室温,再收集反应液分别通过气相色谱-质谱仪和气相色谱进行定性和定量分析,色谱定量分析以邻硝基甲苯为内标物。1,2-二苯乙烷的转化率X以及各产物的选择性Si分别按式(1)和式(2)进行计算[36]。

1.3 分析方法
产物定性分析采用岛津GCMS-QP2010 Plus气质联用仪,色谱柱为DB-5MS毛细管柱(30 m×0.25 mm×0.25 μm)。气相色谱设置条件:进样器290℃;柱箱程序升温(40℃,保持 5 min;10℃·min−1到220℃,保持 2 min;5℃·min−1到 280℃,保持 1 min;5℃·min−1到 290℃,保持 5 min);分流进样(分流比10:1);柱前压49.5 kPa。质谱设置参数:离子源温度200℃,接口温度220℃,检测时间3.2~45 min,扫描速度 1000 amu·s−1,质核比(m/z)范围22~500。溶剂延迟时间3.0 min,离子化检测源。
原料和产物的定量分析采用福立 9790Ⅱ气相色谱分析仪通过内标法进行定量,色谱柱为 FFAP极性毛细管柱(30 m×0.25 mm×0.25 μm),检测器为氢火焰离子化检测器(FID)。色谱分析条件为进样器250℃,检测器280℃,柱箱程序升温(120℃保持 2 min,10℃·min−1升温到 180℃保持 1 min,20℃·min−1升温到 230℃保持 10 min),载气为 N2,柱前压 0.1 MPa,空气 300 ml·min−1,H230 ml·min−1,分流比 50:1,进样量 1 μl。
2 结果与讨论
2.1 NHPI对氧气氧化1,2-二苯乙烷的催化作用
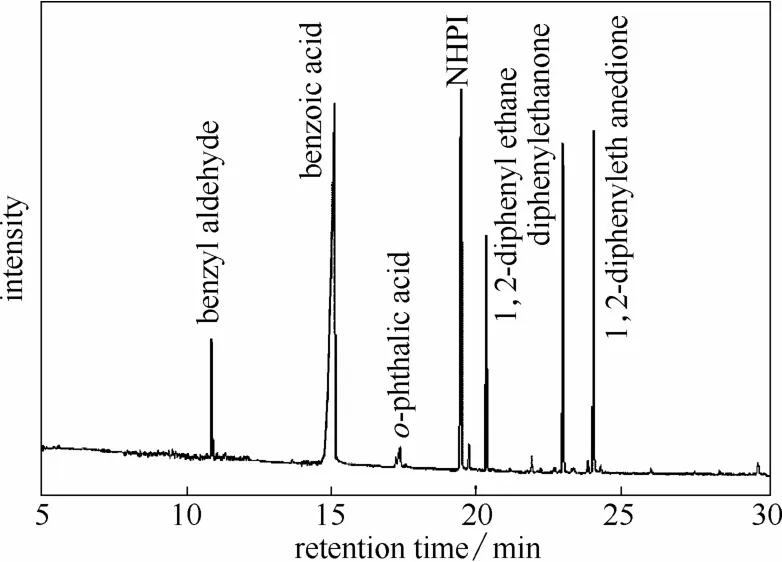
图1 反应产物定性分析结果Fig.1 Products identified by GC-MS

表1 NHPI对氧气氧化1,2-二苯乙烷的催化作用Table 1 Catalytic effect of NHPI on oxidation of 1,2-diphenylethane
在90℃,P(O2)为1.0 MPa,反应时间10 h的条件下考察了NHPI对氧气氧化1,2-二苯乙烷的催化作用,结果如表1所示。当没有催化剂存在时,未检测到氧化产物生成,说明在90℃下1,2-二苯乙烷的自催化氧化过程不能被引发,分子氧不能氧化1,2-二苯乙烷。当加入1,2-二苯乙烷物质的量1%的乙酸锰时,对分子氧氧化 1,2-二苯乙烷仍然未见显著的催化作用。但当向 1,2-二苯乙烷的乙酸溶液中加入1,2-二苯乙烷物质的量10%的NHPI后,反应10 h 1,2-二苯乙烷的转化率为48.6%。GC-MS分析表明反应产物主要为苯甲醛、苯甲酸、二苯基乙酮和二苯乙二酮(图1)。可见NHPI能够催化分子氧氧化1,2-二苯乙烷,氧化反应主要发生在苯环侧链上,其中侧链C—C键断裂产物苯甲醛和苯甲酸的总选择性为31.9%。
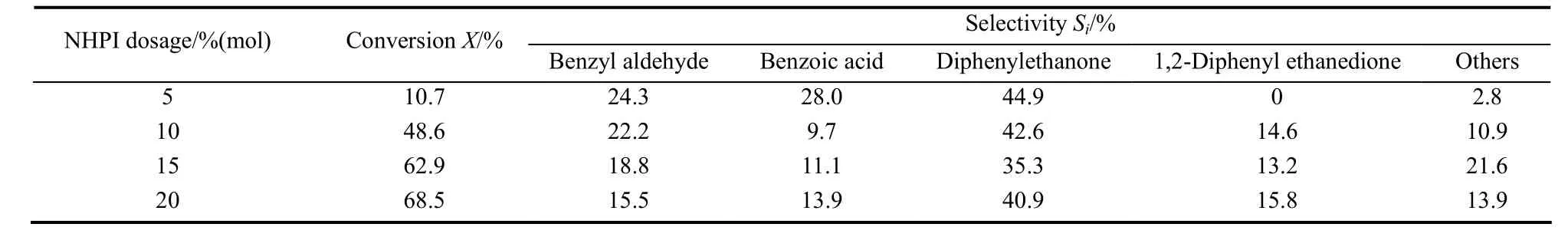
表2 NHPI用量对1,2-二苯乙烷氧化反应的影响Table 2 Effect of NHPI dosage on conversion of 1,2-diphenylethane
2.2 NHPI用量对1,2-二苯乙烷氧化反应的影响
催化剂用量通常对反应速率具有较大的影响,因此进一步考察了NHPI用量对1,2-二苯乙烷反应速率的影响(表2)。从表2可知,1,2-二苯乙烷的氧化速率随着NHPI加入量的增加而增大,当NHPI加入量从 1,2-二苯乙烷物质的量的 5%增大到 10%时,1,2-二苯乙烷的转化率从 10.7%迅速增加到48.6%,而逐步增大 NHPI用量至 20%虽然也能进一步提高 1,2-二苯乙烷的转化速率,但增幅明显减小。同时可以发现进一步增加NHPI用量对产物的选择性影响较小,仅苯甲醛的选择性略有下降,苯甲酸的选择性略有升高。
2.3 不同过渡金属盐的助催化作用
NHPI催化烃类液相氧化的研究发现,Co、Mn等过渡金属的可溶性盐能够促进 NHPI 脱氢生成PINO,从而增强NHPI的催化活性[37]。因此以1,2-二苯乙烷物质的量10%的NHPI为催化剂考察了乙酸铜、乙酸钴、乙酸锰、硝酸铜和硝酸锰作为助剂对NHPI催化氧气氧化1,2-二苯乙烷的助催化作用,结果如表3所示。当分别加入1%1,2-二苯乙烷物质的量的硝酸铜、硝酸锰、乙酸铜、乙酸锰和乙酸钴后,1,2-二苯乙烷的转化率比仅有NHPI时分别提高了15.7%、59.5%、42.6%、70.4%和72.3%。这表明5种过渡金属盐都对NHPI的活性具有很好的促进作用。其中钴盐和锰盐作为助催化剂时C—C键断裂产物苯甲醛和苯甲酸的总选择性较高,均在65%以上。以乙酸锰为助剂时,醛和酸的总选择性为72.7%,比单独使用 1,2-二苯乙烷物质的量 10%的NHPI作为催化剂时提高一倍以上。醛和酸的总收率达到 60.2%。而铜盐的助催化作用比钴盐和锰盐弱,二苯基乙酮的选择性显著高于钴盐和锰盐。这表明过渡金属盐不仅能促进PINO的生成,很可能还直接与氧化反应的底物或某些关键中间产物存在直接作用,从而影响产物选择性。对催化C—C键的氧化断裂而言,乙酸锰是NHPI催化剂最适宜的助剂。
2.4 乙酸锰用量对1,2-二苯乙烷氧化反应的影响
进而考察了乙酸锰用量对 1,2-二苯乙烷转化率和产物选择性的影响(图2)。由图2可知,当NHPI用量为 1,2-二苯乙烷物质的量的 10 %,乙酸锰与NHPI之比为0.05时,1,2-二苯乙烷转化率从48.6%增大到82.9%,苯甲醛和苯甲酸的总选择性从31.9%提高到65.3%。进一步增大乙酸锰与NHPI的比例,对 1,2-二苯乙烷的转化率几乎没有影响。当乙酸锰与NHPI的比值从5%增加到10%时,苯甲酸的选择性略有提高,而二苯基乙酮的选择性则相应下降。虽然提高的乙酸锰与 NHPI的比例并未显著提高1,2-二苯乙烷的转化率,但同样也没有观察到抑制作用。可见乙酸锰与 NHPI存在一个最优比,在NHPI用量为1,2-二苯乙烷物质的量10%时,乙酸锰与NHPI最佳比为1:10。
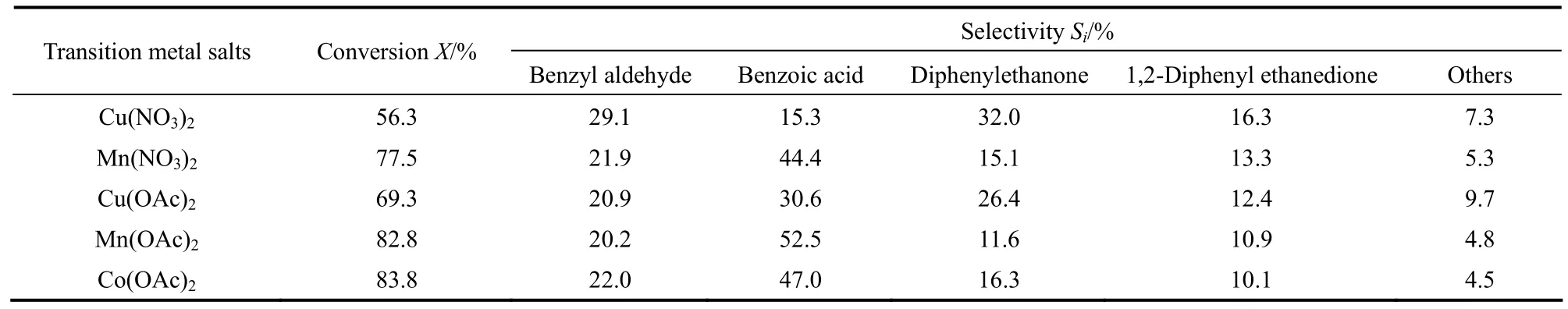
表3 不同过渡金属盐对1,2-二苯乙烷氧化反应的影响Table 3 Promotion effects of various transition metal salts on catalytic activity of NHPI
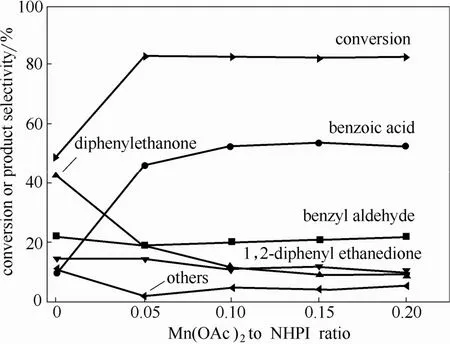
图2 Mn(OAc)2用量对反应速率和产物选择性的影响Fig.2 Effect of Mn(OAc)2amount on 1,2-diphenyl ethane conversion and product selectivity(Reaction conditions:1,2-diphenyl ethane 0.5 g,acetic acid 30.0 g,NPHI amount is 10%( mol) of 1,2-diphneyl ethane,P(O2)=1.0 MPa,T=90℃,reaction time=10 h)
2.5 反应温度对1,2-二苯乙烷氧化反应的影响
不同反应温度下氧气氧化 1,2-二苯乙烷的反应结果如图3所示。当反应温度从70℃升高到80℃底物的转化率显著升高,同时中间产物二苯基乙酮和苯甲醛的选择性显著降低,而苯甲酸的选择性升高。进一步升高反应温度对底物转化率影响甚微,但苯甲酸的选择性增大,二苯基乙酮的选择性继续减小。这说明二苯基乙酮的氧化是 1,2-二苯乙烷氧化中C—C键氧化断裂的关键步骤。可见较高的反应温度对木质素氧化解聚有利,但当反应温度高于 100℃时过度氧化也会变得严重。

图3 反应温度对反应速率和产物选择性的影响Fig.3 Effect of Mn(OAc)2amount on 1,2-diphenyl ethane conversion and product selectivity(Reaction conditions:1,2-diphenyl ethane 0.5 g,acetic acid 30.0 g,NPHI amount is 10%(mol) of 1,2-diphneyl ethane,Mn(OAc)2:NHPI=1:10,P(O2)=1.0 MPa,T=90℃,reaction time=10 h)
2.6 氧气分压对1,2-二苯乙烷氧化反应的影响
不同氧气分压下1,2-二苯乙烷氧化反应的结果如图4所示。从图4可以发现,在所考察的氧压范围内,氧气分压对 1,2-二苯乙烷氧化速率几乎没有影响。这与多数的烃类液相氧化反应一致,底物的氧化反应速率远小于氧气的传质速率,所以宏观动力学上表现出与氧气分压呈0级的反应特征。同时在0.3~2.5 MPa的氧压范围内,也未发现氧气分压对氧化中间产物的分布产生显著的影响。
2.7 1,2-二苯乙烷氧化的分子反应路径

图4 氧气分压对反应速率和产物选择性的影响Fig.4 Effect of oxygen pressure on 1,2-diphenyl ethane conversion and product selectivity(Reaction conditions:1,2-diphenyl ethane 0.5 g,acetic acid 30.0 g,NPHI amount is 10%(mol) of 1,2-diphneyl ethane,Mn(OAc)2:NHPI=1:10,P(O2)=1.0 MPa,T=90℃,reaction time=10 h)
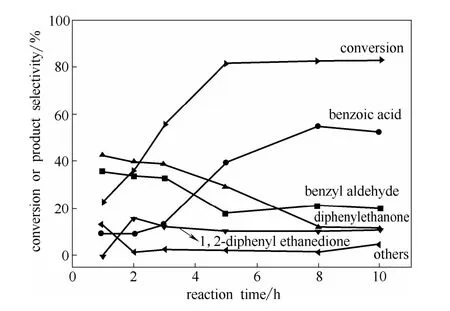
图5 反应时间对反应速率和产物选择性的影响Fig.5 Effect of reaction time on 1,2-diphenyl ethane conversion and product selectivity(Reaction conditions:1,2-diphenyl ethane 0.5 g,acetic acid 30.0 g,NPHI amount is 10%(mol) of 1,2-diphneyl ethane,Mn(OAc)2:NHPI=1:10,P(O2)=1.0 MPa,T=90℃)
以1,2-二苯乙烷物质的量10%的NHPI为催化剂,在Mn(OAc)2:NHPI为1:10,氧气分压1.0 MPa,反应温度90℃的条件下,考察了氧化产物选择性随时间的变化规律(图5)。从图5可以发现,反应初期主要生成大量的二苯基乙酮和苯甲醛,而苯甲酸和二苯乙二酮的选择性较低。这表明二苯基乙酮是氧化反应的初级产物,并且生成速率较快。二苯基乙酮进一步氧化存在两条反应路径,一方面是氧化另一个亚甲基生成二苯乙二酮,另一方面也可以发生C—C键氧化断裂生成两分子的苯甲醛[38]。苯甲醛进一步氧化则生成苯甲酸,而二苯乙二酮进一步氧化也可以生成苯甲酸。而反应的后期主要是二苯基乙酮和苯甲醛的氧化。因此 NHPI和 Mn(OAc)2催化分子氧氧化 1,2-二苯乙烷的反应路径如图6所示。
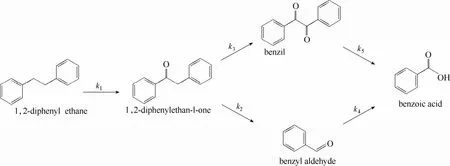
图6 1,2-二苯乙烷氧化反应路径Fig.6 Oxidation pathway of 1,2-diphenyl ethane(Reaction conditions: 1,2-diphenyl ethane 0.5 g,acetic acid 30.0 g,P(O2)=1.0 MPa,T=90℃,reaction time=10 h)
3 结 论
本文以 1,2-二苯乙烷为模型分子研究了 NHPI和过渡金属盐对分子氧选择性氧化断裂木质素分子中C—C键的催化作用。结果表明,NHPI对分子氧氧化1,2-二苯乙烷具有催化作用,Cu、Co和Mn的可溶性盐能够提高NHPI催化活性,其中乙酸锰的助催化作用最强。1,2-二苯乙烷氧化的主要产物是苯甲醛、苯甲酸、二苯基乙酮和二苯乙二酮。提高反应温度、增加乙酸锰用量以及延长反应时间,有利于C—C键的氧化断裂,提高小分子产物的选择性。优化的反应条件为NHPI用量为底物物质的量的10%,乙酸锰和NHPI之比为1:10,反应温度90℃,氧气分压1.0 MPa,反应时间8 h,C—C键的氧化断裂产物苯甲醛和苯甲酸的总选择性可达76.5%。
[1]PAULY M,KEEGSTRA K.Cell-wall carbohydrates and their modification as a resource for biofuels[J].The Plant Journal,2008,54(4): 559-568.
[2]LIU C,WANG H,KARIM A M,et al.Catalytic fast pyrolysis of lignocellulosic biomass[J].Chemical Society Reviews,2014,43(22):7594-7623.
[3]MA R,GUO M,ZHANG X.Selective conversion of biorefinery lignin into dicarboxylic acids[J].ChemSusChem,2014,7(2):412-415.
[4]WYMAN C E,DALE B E,ELANDER R T,et al.Coordinated development of leading biomass pretreatment technologies[J].Bioresource Technology,2005,96(18): 1959-1966.
[5]NIMMANWUDIPONG T,RUNNEBAUM R C,BLOCK D E,et al.Catalytic reactions of guaiacol: reaction network and evidence of oxygen removal in reactions with hydrogen[J].Catalysis Letters,2011,141(6): 779-783.
[6]LI C,ZHAO X,WANG A,et al.Catalytic transformation of lignin for the production of chemicals and fuels[J].Chemical Reviews,2015,115(21): 11559-11624.
[7]CALVO-FLORES F G,DOBADO J,GARC A J I,et al.Lignin and Lignans as Renewable Raw Materials: Chemistry,Technology and Applications[M].West Sussex: John Wiley & Sons,Ltd,2015: 521.
[8]叶跃元.木质素解聚新工艺及机理研究[D].广州: 华南理工大学,2012.YE Y Y.Process and mechanism research on lignin depolymerization[D].Guangzhou: South China University of Technology,2012.
[9]蒋挺大.木质素[M].2版.北京: 化学工业出版社,2009.JIANG T D.Lignin[M].2nd ed.Beijing: Chemical Industry Press,2009.
[10]GELLERSTEDT G.Softwood kraft lignin: raw material for the future[J].Industrial Crops and Products,2015,77: 845-854.
[11]舒日洋,徐莹,张琦,等.木质素催化解聚的研究进展[J].化工学报,2016,67(11): 4523-4532.SHU R Y,XU Y,ZHANG Q,et al.Progress in catalytic depolymerization of lignin[J].CIESC Journal,2016,67(11):4523-4532.
[12]HALMA M,LACHENAL D,MARLIN N,et al.H2O2oxidation of lignin model dimers catalyzed by copper(II)- phenanthroline[J].Industrial Crops and Products,2015,74: 514-522.
[13]OUYANG X P,TAN Y D,QIU X Q.Oxidative degradation of ligninfor producing monophenolic compounds[J].Journal of Fuel Chemistry and Technology,2014,42(6): 677-682.
[14]ZHOU X F.Catalytic oxidation and conversion of kraft lignin into phenolic products using zeolite-encapsulated Cu(II)[H4]salen and[H2]salen complexes[J].Environmental Progress & Sustainable Energy,2015,34(4): 1120-1128.
[15]OUYANG X,RUAN T,QIU X.Effect of solvent on hydrothermal oxidation depolymerization of lignin for the production of monophenolic compounds[J].Fuel Processing Technology,2016,144:181-185.
[16]YANG Q,SHI J,LIN L,et al.Characterization of changes of lignin structure in the processes of cooking with solid alkali and different active oxygen[J].Bioresource Technology,2012,123: 49-54.
[17]RAHIMI A,AZARPIRA A,KIM H,et al.Chemoselective metal-free aerobic alcohol oxidation in lignin[J].Journal of the American Chemical Society,2013,135(17): 6415-6418.
[18]HANSON S K,BAKER R T,GORDON J C,et al.Aerobic oxidation of pinacol by vanadium(V) dipicolinate complexes: evidence for reduction to vanadium(III)[J].Journal of the American Chemical Society,2009,131(2): 428-429.
[19]HANSON S K,BAKER R T,GORDON J C,et al.Mechanism of alcohol oxidation by dipicolinate vanadium(V): unexpected role of pyridine[J].Journal of the American Chemical Society,2010,132(50):17804-17816.
[20]HANSON S K,BAKER R T,GORDON J C,et al.Aerobic oxidation of lignin models using a base metal vanadium catalyst[J].Inorganic Chemistry,2010,49(12): 5611-5618.
[21]HANSON S K,WU R,SILKS L A P.Mild and selective vanadium-catalyzed oxidation of benzylic,allylic,and propargylic alcohols using air[J].Organic Letters,2011,13(8): 1908-1911.
[22]SEDAI B,DÍAZ-URRUTIA C,BAKER R T,et al.Comparison of copper and vanadium homogeneous catalysts for aerobic oxidation of lignin models[J].ACS Catalysis,2011,1(7): 794-804.
[23]HANSON S K,WU R,SILKS L A P.C—C or C—O bond cleavage in a phenolic lignin model compound: selectivity depends on vanadium catalyst[J].Angewandte Chemie International Edition,2012,51(14): 3410-3413.
[24]ZHANG G,SCOTT B L,WU R,et al.Aerobic oxidation reactions catalyzed by vanadium complexes of bis(phenolate) ligands[J].Inorganic Chemistry,2012,51(13): 7354-7361.
[25]SCHEUERMANN M L,LUEDTKE A T,HANSON S K,et al.Reactions of five-coordinate platinum(IV) complexes with molecular oxygen[J].Organometallics,2013,32(17): 4752-4758.
[26]SEDAI B,DÍAZ-URRUTIA C,BAKER R T,et al.Aerobic oxidation of β-1 lignin model compounds with copper and oxovanadium catalysts[J].ACS Catalysis,2013,3(12): 3111-3122.
[27]HANSON S K,BAKER R T.Knocking on wood: base metal complexes as catalysts for selective oxidation of lignin models and extracts[J].Accounts of Chemical Research,2015,48(7): 2037-2048.
[28]DÍAZ-URRUTIA C,SEDAI B,LECKETT K C,et al.Aerobic oxidation of 2-phenoxyethanol lignin model compounds using vanadium and copper catalysts[J].ACS Sustainable Chemistry &Engineering,2016,4(11): 6244-6251.
[29]SHELDON R A,ARENDS I W C E.Catalytic oxidations mediated by metal ions and nitroxyl radicals[J].Journal of Molecular Catalysis A: Chemical,2006,251(1/2): 200-214.
[30]KISHIOKA S Y,YAMADA A.Kinetic study of the catalytic oxidation of benzyl alcohols by phthalimide-n-oxyl radical electrogenerated in acetonitrile using rotating disk electrode voltammetry[J].Journal of Electroanalytical Chemistry,2005,578(1):71-77.
[31]ISHII Y,SAKAGUCHI S,IWAHAMA T.Innovation of hydrocarbon oxidation with molecular oxygen and related reactions[J].Advanced Synthesis & Catalysis,2001,343(5): 393-427.
[32]ISHII Y,SAKAGUCHI S.Recent progress in aerobic oxidation of hydrocarbons byn-hydroxyimides[J].Catalysis Today,2006,117(1/2/3): 105-113.
[33]IWAHAMA T,SYOJYO K,SAKAGUCHI S,et al.Direct conversion of cyclohexane into adipic acid with molecular oxygen catalyzed byn-hydroxyphthalimide combined with Mn(acac)2and Co(OAc)2[J].Organic Process Research & Development,1998,2(4): 255-260.
[34]SHIRAISHI T,TAKANO T,KAMITAKAHARA H,et al.Studies on electro-oxidation of lignin and lignin model compounds(Part 2):N-hydroxyphthalimide (NHPI)-mediated indirect electro-oxidation of non-phenolic lignin model compounds[J]. Holzforschung:International Journal of the Biology,Chemistry,Physics,&Technology of Wood,2012,66(3): 311-315.
[35]PINHEIRO F G C,SOARES A K L,SANTAELLA S T,et al.Optimization of the acetosolv extraction of lignin from sugarcane bagasse for phenolic resin production[J].Industrial Crops and Products,2017,96: 80-90.
[36]GÜVENATAM B,HEERES E H J,PIDKO E A,et al.Decomposition of lignin model compounds by lewis acid catalysts in water and ethanol[J].Journal of Molecular Catalysis A: Chemical,2015,410:89-99.
[37]ISHII Y,IWAHAMA T,SAKAGUCHI S,et al.Alkane oxidation with molecular oxygen using a new efficient catalytic system:N-hydroxyphthalimide (NHPI) combined with Co(acac)n(n=2 or 3)[J].The Journal of Organic Chemistry,1996,61(14): 4520-4526.
[38]ZHOU Y,LIN S,BIAN Y,et al.Aerobic oxidation of aromatics catalyzed by CoSPc and NHPI[J].Indian Journal of Chemistry -Section B Organic and Medicinal Chemistry,2016,55B(5): 624-628.
Oxidative C—C bond cleavage of lignin model compound with O2in presence ofN-hydroxyl phthalimide
LIU Changjun1,CHEN Ning1,LIU Yingying2,LU Houfang1,LIANG Bin1,2,TANG Siyang1,YUE Hairong1,TAN Pinghua3
(1School of Chemical Engineering,Sichuan University,Chengdu610207,Sichuan,China;
2Center of CCUS and CO2Mineralization and Utilization,Sichuan University,Chengdu610207,Sichuan,China;
3Southwest Research & Design Institute of Chemical Industry,Chengdu610225,Sichuan,China)
O 643.32; O 625
A
0438—1157(2017)10—3788—07
10.11949/j.issn.0438-1157.20170533
2017-05-02收到初稿,2017-06-28收到修改稿。
联系人:刘长军,谭平华。
刘长军(1979—),男,博士,副教授。
国家自然科学基金项目(21406146,21476150,21336008)。
Received date:2017-05-02.
Corresponding author:LIU Changjun,liuchangjun@scu.edu.cn; TAN Pinghua,kinghuatan@163.com
Foundation item:supported by the National Natural Science Foundation of China(21406146,21476150,21336008).
