蛋白质与RNA相互作用的预测和研究
2017-10-13薛建新
王 彤,薛建新,杜 奕
(上海第二工业大学计算机与信息工程学院,上海201209)
蛋白质与RNA相互作用的预测和研究
王 彤,薛建新,杜 奕
(上海第二工业大学计算机与信息工程学院,上海201209)
确定蛋白质与RNA是否发生作用非常重要,因为它广泛存在于生物学过程中,在生物体细胞活动中起到至关重要的作用。特别是近几年随着蛋白质结构数据的增多,如果仍用传统的物理化学方法去测定会非常困难,找到能自动预测蛋白质与RNA的相互作用的方法迫在眉睫。首先采用PsePSSM算法表达蛋白质序列,编码后的蛋白质特征向量维数很高;接着采用GPP流形学习方法对其进行维数约简,约简后的特征向量输入SVM分类器训练,训练好的分类器预测未知的蛋白质与RNA是否相互作用;最后,采用Jackknife测试方法检验预测准确率,测试结果表明,上述方法是十分有效的,为蛋白质与RNA是否相互作用的研究提供一条新的思路。
蛋白质与RNA相互作用;维数约简;预测
0 引言
蛋白质与RNA的相互作用很重要,可以体现出蛋白质的功能。同时,细胞内各种重要的生理过程,以蛋白质与RNA的相互作用为基础,这些生理过程包括信号的转导等。蛋白质与RNA的相互作用在蛋白合成[1]、病毒复制[2-3]和转录调控[4]等方面都有广泛的应用。因此,在生物信息学中,蛋白质与RNA的相互作用的研究占有很重要的地位。
如今,许多蛋白质与RNA复合物的三维结构被测出。如果采用传统的实验方法测定这些生物数据会带来很多问题,如成本高、耗时长等。因此,提出采用机器学习算法来预测蛋白质与RNA是否相互作用,可以有效地解决传统实验方法带来的问题。Liu等[5]采用氨基酸序列和结构描述子编码蛋白质序列,然后采用随机森林算法来预测蛋白质与RNA是否相互作用。Kumar等[6]采用氨基酸组成和进化信息编码蛋白质序列,然后采用支持向量机(Support Vector Machines,SVM)来区分蛋白质与RNA是否相互作用,但不能预测未知蛋白质与RNA是否相互作用。
本文提出的方法解决了上述的问题。首先,采用伪特定位点记分矩阵(Pseudo Position-Specifi c Scoring Matrix,PsePSSM)[7]序列编码方法来表示蛋白质与RNA序列对。将这种特征提取方法引入到蛋白质与RNA相互作用的预测问题中,能显著提高预测准确率。因为采用这种特征提取方法编码的蛋白质与RNA序列对,包含了蛋白质序列的相似度和进化信息。它尽可能多地保留了蛋白质序列的原始信息,但同时它可能会导致小样本问题。为了解决这个问题,本文采用新的降维(Dimensionality Reduction,DR)算法从原始高维向量中提取关键特征向量。它有2个主要的降维技术:过滤和包裹。过滤技术与分类算法无关,与包裹方法相比,其优点是计算简单快速,所以很容易应用到非常高维数据集,如本文的研究中所使用的特征向量。然后,基于降维后的低维特征向量,SVM分类器预测蛋白质与RNA的相互作用。实验结果表明,本文的方法是非常有效的,为蛋白质与RNA是否相互作用的研究提供一条新的思路。
1 材料和方法
1.1 数据集
建立一个数据集。首先,本文检索了603个RNA结合蛋白复合物,以X射线结晶分析的分辨率≤0.35 nm进行过滤。去除序列同源性上分别大于25%的蛋白质和RNA链,得到了365个非冗余蛋白RNA链。对于负样本集合,从UniProt数据库中收集了200个不与RNA相互作用的蛋白质序列。
给定一个待查询蛋白质与RNA序列对P,预测它是否相互作用,需要做的第1件重要的事情是采用适当的编码方法来表达它,序列编码方法PsePSSM就是其中一种。
1.2 PsePSSM序列编码方法
蛋白质由20种氨基酸组成,每条蛋白质序列用字符串表示,首先将蛋白质字符序列离散成数值序列。本文采用PsePSSM编码方式。下面介绍PsePSSM[7]方法。
依据参考文献[7],PsePSSM矩阵可以表示为

式中,
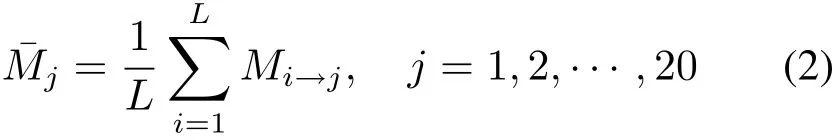
L为某蛋白质P的长度,式(2)中的分数Mi→j表示该蛋白质序列P的第i-th个位置的氨基酸突变成第j种氨基酸的得分。序号1,2,···,20用来表示20种氨基酸中的一种(按照字母顺序表排列)。利用PSI-BLAST程序搜索Swiss-Prot得到Mi→j[8]。其中用于PSI-BLAST的参数为:3次循环,E值为0.001。根据PSSM的定义,用如下L×20的分数矩阵表示蛋白质序列P[7]。
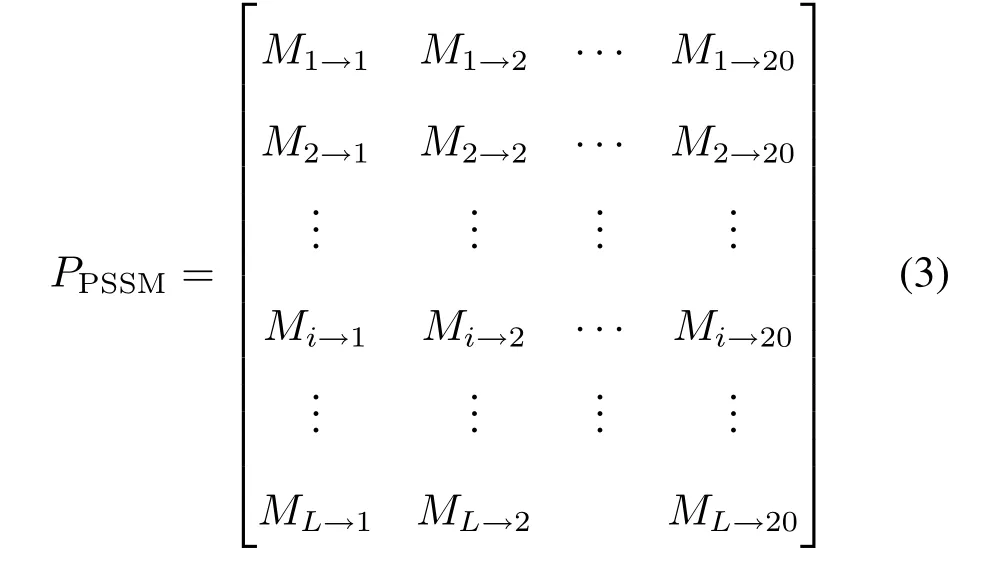
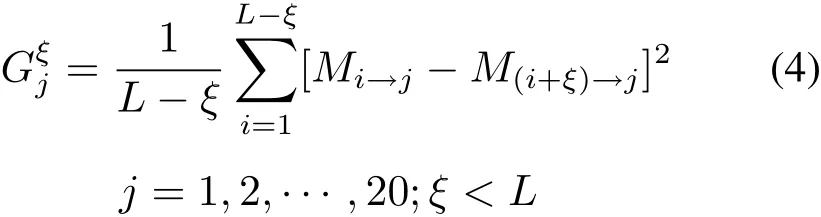
根据式(1),1条蛋白质序列表示为1个高维向量包含1个20维的向量(ξ=0)和49个40维的向量 (ξ=1,2,···,或 49)。这 49 个 40 维的向量中,中前20维的向量都是相同的,去掉重复的向量,保留1个20维向量。得到1个1 000(20+49×20)维的向量。1 000维的高维特征向量会使预测系统复杂化。这里,引入GPP方法来解决这个难题。
1.3 GPP降维算法
几何保留投影(Geometry Preserving Projections,GPP)[9]是一种线性降维算法。GPP的思想是保留局部的信息,通过捕捉特征空间类间的几何属性和类内的几何性质来实现降维。对于关于GPP的概念更详细的描述,参见文献[9]。下面,简要介绍一下GPP。
数 据 集 X = [→x1,→x2,···,→xN] 是 由 m- 维的实数空间Rm内给出的,数据集包含C个类 别 [Φ1,Φ2,···,ΦC], 并 且 每 个 数 据 点 →xi(i=1,2,···,N)分别属于某一个类别。算法将原始数据X通过投影矩阵投影到低维空间Rd(d<m)。目标是找到最优的投影矩阵B:

为了得到最优的投影矩阵B,需最小化如下的目标函数:
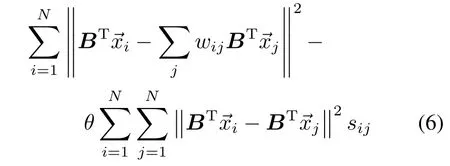
式中,wij(i,j=1,2,···,N)为系数矩阵,sij(i,j=1,2,···,N)为相似度矩阵。θ为尺度因子,其范围是[0,1]。
想要最小化目标函数,即等价于最小化其被减数和最大化其减数:
(1)最大化其减数,即
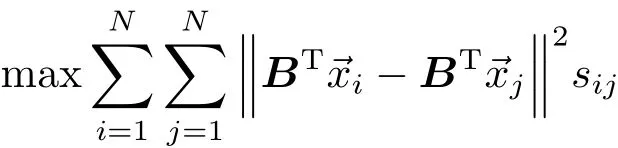
满足条件:

(2)最小化其被减数,即
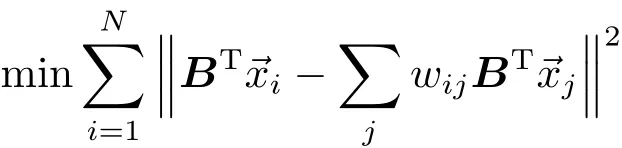
如果 →xi和 →xj属于不同的类别,wij=0,并且
算法计算的样本点都标记过类别信息,不包含未标记的样本子集
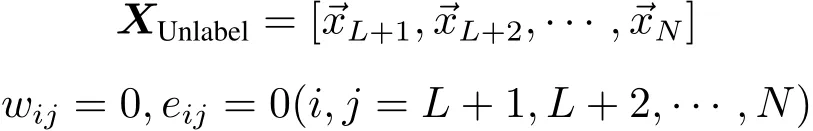
式(6)可以简化为:
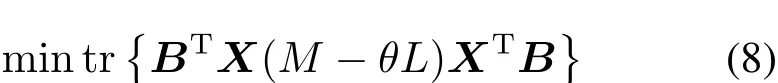
min trBTX(M −θL)XTB (8)
为了能唯一地确定矩阵B,施加约束BTB=I,也就是,B的列向量之间是正交的。现在,目标函数可以写成如下的形式:
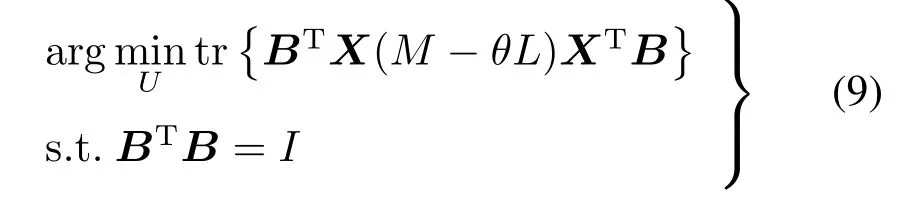
上述问题转化为特征值求解问题:

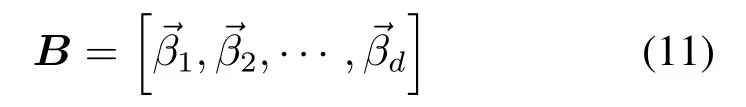
上述的GPP算法能有效地避免小样本问题,因为它不同于以往的算法,GPP算法没有矩阵的逆运算,避免了受到奇异值问题的困扰。
2 结果与讨论
用GPP算法针对1 000-D特征向量降维,最终得到70-D特征向量。分别输入SVM和K近邻(K Nearest Neighbor,KNN)分类器进行训练,训练好的分类器用来预测蛋白质与RNA是否相互作用。
采用Jackknife测试本文提出方法的准确率[10]。为便于比较,没有采用GPP算法得出的结果也列在表1中。从表1可以看出,采用GPP降维算法和SVM分类器后的Jackknife测试可以获得超过98%的准确率,这比没有采用GPP算法得到的准确率高约5%。实验结果表明,通过降维冗余信息被去掉了,同时原始数据中有用的信息被保留了下来。所以预测系统得到了简化,同时分类准确率还提高了。原始向量由1 000维降到了70维,小于样本数,小样本问题也得到了解决。

表1 采用不同的方法预测蛋白质与RNA的相互作用的Jackknife准确率Tab.1 The Jackknifesuccessratesfor protein-RNA interaction prediction by different methods
此外,需调整KNN分类器中的最近邻数K,K的取值大小会影响分类的性能。基于不同K下的预测准确率如图1所示。可以看出,当K=1时采用GPP算法得出的预测准确率为最大值,不采用GPP算法时,预测准确率最大值也在K=1时取得。
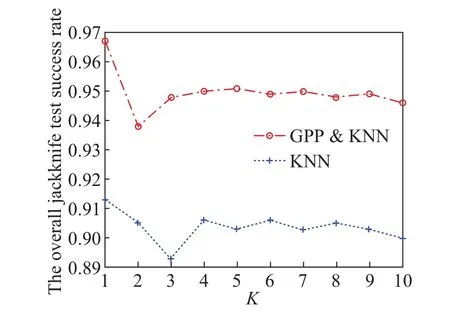
图1 采用KNN方法当K取不同值时的Jackknife预测准确率比较结果Fig.1 The comparison results of Jackknife prediction success rates obtained by KNN algorithms with different K
3 结语
本文所提出的方法在预测蛋白质与RNA相互作用方面是非常有效的,现有的预测主要集中在寻找最佳的分类方案,笔者则是从另外一个角度考虑简化生物系统的复杂性。本文应用GPP算法从高维空间中提取关键信息,同时还解决了小样本问题,基于降维后的特征向量利用SVM来预测蛋白质与RNA是否相互作用。结果表明,该方法降低了预测系统的复杂性,解决了小样本问题,同时还提高了预测的准确率。
[1] BEAUDOINM E,POIREL V J,KRUSHEL L A.Regulating amyloid precursor protein synthesisthrough an intemal ribosomal entry site[J].Nucleic Acids Res,2008,36(21):6835-6847.
[2] NEWCOMBL L,KUORL,YEQ,etal.Interaction of the infl uenza a virus nucleocapsid protein with the viral RNA polymerasepotentiatesunpfimed viral RNA replication[J].JVirol,2009,83(1):29-36.
[3] YU Z,SANCHEZ-VELAR N,CATRINA I E,et al.The cellular HI V-1 Rev cofactor hRIP is required for viral replication[J].Proc Natl Acad Sci USA,2005,102(11):4027-4032.
[4]ABDELMOHSEN K,KUWANO Y,KIM H H,et al.Posttranscriptional gene regulation by RNA-binding proteins during oxidative stress:Implications for cellular senescence[J].Biol Chem,2008,389(3):243-255.
[5] LIU ZP,MIAOH.Prediction of protein-RNA interactions using sequenceand structuredescriptors[J].Ifac Papersonline,2016,48(28):28-34.
[6]KUMAR M,GROMIHA M M,RAGHAVA G P.SVM based prediction of RNA-binding proteins using binding residues and evolutionary information[J].JMol Recognit,2011,24(2):303-313.
[7] CHOU K C,SHEN H B.MemType-2L:A web server for predicting membrane proteins and their types by incorporating evolution information through Pse-PSSM[J].Biochemical and Biophysical Research Communications,2007,360(2):339-345.
[8] 王彤,薛建新,孔亮亮.细菌性病原体内病毒蛋白的预测和研究[J].上海第二工业大学学报,2016,33(3):231-235.
[9] ZHANG T H,LI X L,TAO D C,et al.Multimodal biometrics using geometry preserving projections[J].Pattern Recognition,2008,41(3):805-813.
[10]王彤,薛建新,谭文安.利用半监督降维算法预测蛋白质亚细胞位置[J].上海第二工业大学学报,2015,32(3):260-265.
Abstract:It is very important to determine whether RNA and protein interacts or not.Because it is widely present in the biological process and plays a vital role in the biological cell activity.Especially,in recent years,with the increase of protein structure data,it is very diffi cult to determine the interaction between protein and RNA with traditional physical and chemical methods.It is imminent to fi nding a way to predict the interaction between proteins and RNA.Firstly,the PsePSSM algorithm was used to express the protein sequence.The feature vector dimension of the encoded protein was very high.Then the GPPmanifold learning method was used to reduce the dimension of the protein.The reduced feature vector was input into the SVM classifi er,and the trained classifi er predictors wereused to predict whether theunknown protein interacted with the RNA.Finally,the Jackknifemethod wasused to test theaccuracy of prediction.The resultsshowed that themethod wasvery effective.It can providea new way to study theinteraction between protein and RNA.
Keywords:interaction between protein and RNA;dimensionality reduction;prediction
The Prediction and Research of RNA-Protein Interactions
WANGTong,XUEJianxin,DU Yi
(School of Computer and Information Engineering,Shanghai Polytechnic University,Shanghai201209,China)
TP 391;Q 617
A
1001-4543(2017)03-0227-04
10.19570/j.cnki.jsspu.2017.03.011
2017-04-10
王 彤(1981–),女,河北保定人,副教授,博士,主要研究方向为数据挖掘算法及其应用。E-mail:wangtong@sspu.edu.cn。
国家自然科学基金(No.61672022,No.61502296),上海市自然科学基金(15ZR1417000)资助
