具除草活性的羟基唑并嘧啶的合成和评价
2017-10-13章乐天编译
章乐天 编译
具除草活性的羟基唑并嘧啶的合成和评价
章乐天 编译
(华东理工大学 药学院,上海 200237)
用除草剂来控制作物田的杂草种群是一项成功的技术,在2014年的传统作物保护市场560亿美元中除草剂占43%。监管和除草剂抗性问题给除草剂的使用带来了挑战,例如对已知作用机制的非选择性和/或选择性除草剂抗性杂草继续增加。需要许多策略来解决这些问题,如开发和发现新作用机制的除草剂。最近,巴斯夫进行了高通量筛选,确定在异戊二烯生物合成的非甲羟戊酸途径中发现一系列先导结构,并得到酶抑制剂。几种类型的化合物抑制途径中的第三种酶,即4-二磷酸胞苷2C-甲基--赤藓糖醇合酶(IspD)最有效的抑制剂是6-苄基-取代的羟基唑并嘧啶1(图1)。
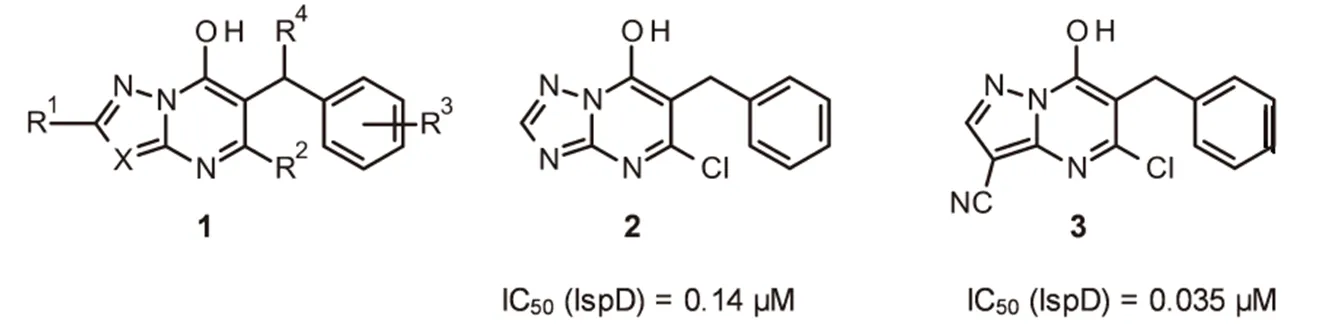
图1 已报道的IspD的抑制剂
羟基三唑并嘧啶2被确定为亚微摩尔抑制剂,研究表明其在蛋白质的变构位点结合IspD。化合物2和IspD的共晶体结构允许优化结合的配体,由此得到了羟基吡唑并嘧啶3,其活性提高了4倍。即使在高浓度(有效成分3 kg/hm2)时,在生测试验中,该系列1具有白化作用,化合物2具有优异的活性。另外,在叶碟生测试验中,当以100 μM的浓度施用时,化合物2和3对番茄红素积累有>60%的抑制作用。据报道,苄基位置(R4)的取代降低了系列1的结合亲和力;而右侧环(R3)的取代是可以接受的,这导致设计了系列化合物4,其中唑并嘧啶核心和苯环通过醚氧键连接,目的是发现活体活性增加的化合物,其也可以是IspD的抑制剂(图2)
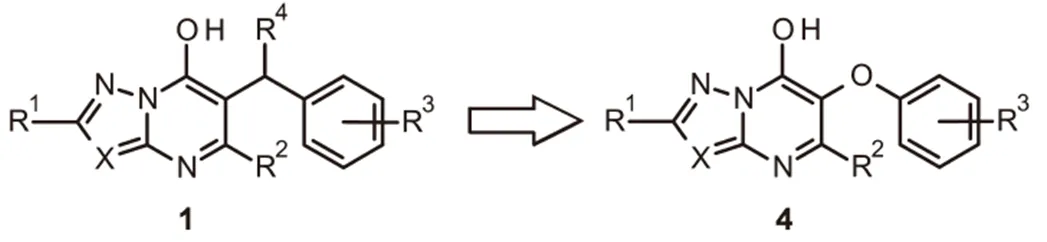
芳氧基系列
1 材料和方法
1.1 化学合成
1.1.1 通用方法
合成化合物12-14和31以及三唑并嘧啶61-63所需苯酚的合成以及所有新化合物的核磁检测法用已有的方法。氢核磁共振谱检测用Bruker Avance 3 HD400(400MHz)或Bruker Avance 3 500(500MHz)光谱仪(Bruker,Billerica,MA),以CDCl3或DMSO-d6为溶剂。化学位移以保留2位小数的数字表示,单位ppm,信号分裂记录为单峰(s)、双峰(d)、三重峰(t)、四重峰(q)、五重峰(quin)、六分图(sxt)、七重峰(spt)和多重峰(m)。耦合常数J保留一位小数,单位为Hz。用Bruker Avance 3 500核磁共振仪以125MHz测定碳谱,以CDCl3或DMSO-d6为参照物。化学位移保留一位小数,单位ppm。低分辨质谱(LRMS)采用沃特斯的超高效液相色谱Acquity UPLC(Waters Corp.,Milford,MA)检测,配置沃特斯SQD2单四极杆MS或岛津QP2010+单四极杆GC-MS(Shimadzu Corp.,Kyoto,Japan)。高分辨质谱(HRMS)采用沃特斯的Acquity UPLC检测,配置赛默飞Orbitrap XL (Thermo Fisher Scientific,Waltham,MA)。其值为质荷比,单位道尔顿。使用未校正Optimelt自动熔点系统(Standford Research Systems,Sunnyvale,CA)测定熔点。用质量定向的制备型HPLC进行纯化,Waters Fraction Lynx系统用UV与ES +的混合触发剂,该系统包括具有2525梯度泵的2767注射器/收集器、泵控制模块、2个515等度泵、CFO、2996光电二极管阵列、2420 蒸发光散射检测器(ELSD)和Micromass ZQ2000。使用沃特斯Atlantis dC185 μm 19×10 mm 保护柱和沃特斯 Atlantis dC185 μm OBD 30×100 mm制备柱。柱洗脱液为H2O(含有0.05%TFA)和乙腈(含有0.05%TFA)的梯度混合液。
1.1.2 一般方法1——合成2-芳氧基丙二酸酯
在通入氮气的情况下,将钠(1.01当量)分批加入到乙醇(1.0 mL/mmol)中。将混合物搅拌2 h,直到金属钠完全溶解。加入相应的苯酚(1.0当量),搅拌混合物10 min。滴加氯代丙二酸二乙酯9(1.2当量),将所得溶液加热回流16 h。将混合物冷却至室温并真空浓缩。将残留物悬浮于水(0.5 mL/mmol)中,用Et2O(3×0.5 mL/mmol)萃取。合并有机萃取物,用水(0.5 mL/mmol)冲洗,然后用MgSO4干燥,过滤并真空浓缩。用快速柱色谱纯化粗产物,得到相应的2-芳氧基丙二酸酯。
1.1.3 2-苯氧基丙二酸二乙酯的合成(10)
在一般方法1中加入苯酚(8)。快速柱色谱法(乙酸乙酯︰异己烷=1︰19)纯化粗产物,得到无色油状物(1.53 g),通过Kugelrohr蒸馏进一步纯化,得到无色的丙二酸酯10油状物(900 mg,34%)。LRMS:275(M+Na+);1HNMR(400 MHz,CDCl3)δH:7.24-7.35 (2H,m),7.04(1H,t,J=7.4),6.95-7.00(2H,m),5.21(1H,s),4.25-4.39(4H,m),1.31(6H,t,J=7.1)。
1.1.4 2-(2,6-二氯苯氧基)丙二酸二乙酯(11)的合成
一般方法1中加入2,6-二氯苯酚(10.0 g)。快速柱色谱法纯化粗产物(乙酸乙酯︰异己烷=1︰19),得到无色油状的丙二酸酯11(16.3 g,83%)。LRMS:(321,M+H+);1H NMR(400 MHz,CDCl3)δH:7.30(2H,d,J=8.1),6.99-7.08(1H,m),5.14(1H,s),4.25-4.39 (4H,m),1.31(6H,t,J=7.1)。
1.1.5 2-(3-溴-2-氯-6-氟-苯氧基)丙二酸二乙酯(12)的合成
在一般方法1中加入3-溴-2-氯-6-氟苯酚(5.0 g,22.2 mmol)。快速柱色谱法(乙酸乙酯︰异己烷=1︰9)纯化粗产物,得到无色油状的化合物12(7.14 g,84%)。GCMS:(384,M+);1H NMR(400 MHz,CDCl3) δH:7.38(1H,dd,J=9.1和5.0),6.96(1H,dd,J=10.3和9.1),5.21(1H,s),4.26-4.41(4H,m),1.32(6H,t,J=7.1)。
1.1.6 2-[2,6-二氯-3-(三氟甲基)苯氧基]丙二酸二乙酯(13)的合成
在一般方法1中加入2,6-二氯-3-(三氟甲基)苯酚(1.5 g,6.2 mmol)。快速柱色谱法(乙酸乙酯︰异己烷=1︰9)纯化粗产物得到无色油状的化合物13 (1.8 g,75%)。GCMS:(389,M+);1H NMR(400 MHz,CDCl3)δH:7.40-7.51(2H,m),5.18(1H,s),4.26-4.41 (4H,m),1.32(6H,t,J=7.1)。
1.1.7 2-[6-氯-2-氟-3-(三氟甲基)苯氧基]丙二酸二乙酯的合成(14)
在一般方法1中加入6-氯-2-氟-3-(三氟甲基)苯酚(3.4 g,15.8 mmol)。快速柱色谱法(乙酸乙酯︰异己烷=1︰9)纯化粗产物得到无色油状物,经Kugelrohr蒸馏进一步纯化,得到无色油状的化合物14 (2.9 g,49%)。GCMS:(372,M+);1H NMR(400 MHz,CDCl3)δH:7.37-7.28(2H,m),5.25(1H,s),4.41-4.27 (4H,m),1.32(6H,t,J=7.1)。
1.1.8 一般方法2-两步方法制备5,7-二氯-6-[芳氧基]-[1,2,4]三唑并[1,5-a]嘧啶
在氮气保护下将钠(1.05当量)加入乙醇(3.0 mL/mmol)中。搅拌混合物1 h,直到金属钠完全溶解。加入相应的丙二酸二乙酯(1.0当量)和3-氨基-1,2,4-三唑15(1.0当量),将混合物加热回流18 h。将混合物冷却至室温并真空浓缩。将残留物溶于H2O(5.0 mL/mmol)中。点滴加入HCl水溶液(2.0 M),将混合物的pH调至1,并过滤沉淀物。将滤液在真空中干燥,得到相应的二酮,悬浮于磷酰氯(10 mL/g)中。将混合物加热回流(110 ℃)6 h,然后冷却至室温。将混合物小心地倒入0 ℃的冰水混合物(10 g/g)中。混合物用CH2Cl2(3×20 mL/g)萃取,合并的有机萃取物,用MgSO4干燥,过滤并真空浓缩。快速柱色谱纯化粗产物,得到相应的5,7-二氯-6-[芳氧基]-[1,2,4]三唑并[1,5-a]嘧啶。
1.1.9 5,7-二氯-6-苯氧基-[1,2,4]三唑并[1,5-a]嘧啶(17)的合成
在一般方法2中加入化合物10(6.5 g,25.8 mmol)制备产物。快速柱色谱法(乙酸乙酯︰异己烷=2︰3)纯化粗产物,得到浅黄色固体化合物17(3.8 g,2步最终产率大于63%)。LRMS:(283,M+H+);1H NMR (400MHz,CDCl3)δH:8.59(1H,s),7.33-7.42(2H,m),7.17(1H,t,J= 7.5),6.89-6.95(2H,m)。
1.1.10 5,7-二氯-6-(2,6-二氯苯氧基)-[1,2,4]三唑并[1,5-a]嘧啶(20)的合成
在一般方法2中加入化合物11(1.46 g,4.55 mmol)进行制备产物。快速柱色谱法(乙酸乙酯︰异己烷=2︰3)纯化粗产物,得到白色固体三唑并嘧啶20(0.43 g,2步产率32%)。(LRMS):(353,M+H+);1H NMR(400 MHz,CDCl3)δH:8.59(1H,s),7.40(2H,d,J=8.1),7.14-7.21 (1H,m)。
1.1.11 6-(3-溴-2-氯-6-氟-苯氧基)-5,7-二氯-[1,2,4]三唑并[1,5-a]嘧啶(21)的合成
在一般方法2中加入化合物12(3.0 g,7.82 mmol)进行制备产物。快速柱色谱法(乙酸乙酯︰异己烷=2︰3)纯化粗产物,得到浅黄色固体三唑并嘧啶21,为(0.99 g,2步产率31%)。LRMS:(413,M+H+);1H NMR(400 MHz,CDCl3)δH:8.57(1H,s),7.49(1H,dd,J=9.1和5.0),7.00(1H,dd,J=10.7和9.0)。
1.1.12 5,7-二氯-6-[2,6-二氯-3-(三氟甲基)苯氧基]-[1,2,4]三唑并[1,5-a]嘧啶(22)的合成
在一般方法2中加入化合物13(1.6 g,4.11 mmol)制备产物。快速柱色谱法(乙酸乙酯︰异己烷=2︰3)纯化粗产物,得到无色油状的三唑并嘧啶22(0.27 g,2步产率16%)。LRMS:(419,M+H+);1H NMR(400 MHz,CDCl3)δH:8.57(1H,s),7.63-7.55(1H,m),7.54-7.48 (1H,m)。
1.1.13 5,7-二氯-6-[6-氯-2-氟-3-(三氟甲基)苯氧基]-[1,2,4]三唑并[1,5-a]嘧啶(23)的合成
在一般方法2中加入化合物14(2.87 g,7.70 mmol)制备产物,粗产物通过快速柱色谱法(乙酸乙酯︰异己烷=2︰3)纯化,得到白色泡沫状的三唑并嘧啶23 (0.38 g,2步产率12%)。LRMS:(403,M+H+);1H NMR (400 MHz,CDCl3)δH:8.60(1H,s),7.49-7.39(2H,m)。
1.1.14 一般方法3-5,7-二氯-6-[芳氧基]-[1,2,4]三唑并[1,5-a]嘧啶与氢氧化钠水溶液部分水解反应
把NaOH水溶液(2.0 M,2.0当量)加入5,7-二氯-6-[芳氧基]-[1,2,4]三唑并[1,5-a]嘧啶(1.0当量)的四氢呋喃(5.0 mL/mmol)溶液中。将混合物在回流下搅拌6 h。将反应液冷却至室温,然后加入HCl水溶液(2.0 M)把pH调至1。将混合物用乙酸乙酯(5.0 mL/mmol)稀释,然后萃取分离,用MgSO4干燥有机相,过滤并真空浓缩,得到5-OH和7-OH异构体混合物。用制备型HPLC获得所需的7-OH产物,即5-氯-6-芳氧基-[1,2,4]三唑并[1,5-a]嘧啶-7-醇。
1.1.15 5-氯-6-苯氧基-[1,2,4]三唑并[1,5-a]嘧啶-7-醇(18)的合成
在一般方法3中加入化合物17(0.70 g,2.49 mmol)制备产物,得到异构体(3︰1)混合物。用制备型HPLC提纯得到白色固体18(99 mg)以及化合物19︰18为7︰1的混合物。过滤萃取水溶液获得其余的化合物18。合并,得到白色固体化合物18(0.39 g,61%)和白色固体化合物19(66 mg,10%)。通过X射线晶体学分析测定产物的区域选择性。LRMS:(263M+H+);1H NMR(400MHz,DMSO-d6)δH:8.93(1H,s),7.26-7.33 (2H,m),7.03(1H,t,J=7.3),6.95-7.00(2H,m);mp:289~296 ℃。
1.1.16 7-氯-6-苯氧基-[1,2,4]三唑并[1,5-a]嘧啶-5-醇(19)的分析
LRMS:(263M+H+);1H NMR(400MHz,DMSO-d6) δH:(7︰1混合物19︰18中化合物19):8.24(1H,s),7.28-7.35(2H,m),7.11-7.17(2H,m),7.04-7.10 (1H,m)。
1.1.17 5-氯-6-(2,6-二氯苯氧基-[1,2,4]三唑并[1,5-a]嘧啶-7-醇(24)的合成
在一般方法3中加入化合物20(0.42 g,1.20 mmol)制备产物,得到初始的异构体7︰1混合物。制备型HPLC提纯得到白色固体化合物24(46 mg,12%)。LRMS:(331,M+H+);1H NMR(400 MHz,DMSO-d6) δH:8.88(1H,s),7.54-7.39(2H,m),7.23-7.11(1H,m)。
1.1.18 6-(3-溴-2-氯-6-氟苯氧基)-5-氯-[1,2,4]三唑并[1,5-a]嘧啶-7-醇(25)的合成
在一般方法3中加入化合物21(0.92 g,2.25 mmol)制备产物,得到初始的异构体7︰1混合物。制备型HPLC提纯得到白色固体化合物25(0.10 g,32%)。LRMS:(395,M+H+);1H NMR(400 MHz,DMSO-d6) δH:8.89(1H,s),7.59(1H,dd,J=9.0和5.0),7.29(1H,dd,J=11.3和9.0);mp:291~293 ℃。
1.1.19 合成5-氯-6-[2,6-二氯-3-(三氟甲基)苯氧基]-[1,2,4]三唑并[1,5-a]嘧啶-7-醇(26)
在一般方法3中加入化合物22(0.20 g,0.49 mmol)制备产物,得到10︰1的初始异构体的混合物。制备型HPLC提纯得到白色固体化合物26(32 mg,16%)。LRMS:(401,M+H+,100%);1H NMR(400 MHz,DMSO-d6)δH:8.84(1H,s),7.74-7.64(2H,m);mp:264~269 ℃。
1.1.20 5-氯-6-[6-氯-2-氟-3-(三氟甲基)苯氧基]-[1,2,4]三唑并[1,5-a]嘧啶-7-醇(27)
在一般方法3中加入化合物23(0.36 g,0.87 mmol)制备产物,得到42︰1异构体混合物,即产物27(0.26 g,76%),未进一步纯化即使用。LRMS:(383,M+H+);1H NMR(400 MHz,DMSO-d6)δH:8.94(1H,s),7.64-7.53(2H,m)。
1.1.21 3-氧代-2-苯氧基-丁酸乙酯的合成(29)
在0 ℃下,将氢化钠(0.42 g,10.6 mmol,矿物油中60%分散体)加入苯酚8(1.0 g,10.6 mmol)的四氢呋喃(120 mL)溶液中。搅拌混合物30 min,加入2-氯-3-氧代丁酸乙酯28(2.1 mL,14.8 mmol)中。将混合物温加热至室温并搅拌1 h。将反应混合物加热回流8 h,然后冷却至室温。用Et2O(10 mL)稀释混合物,然后用H2O(10 mL)洗涤。用Et2O(2×10 mL)萃取水层,合并有机萃取液并用H2O(10 mL)洗涤,然后用MgSO4干燥。过滤并真空浓缩。用柱色谱(乙酸乙酯︰异己烷=1︰19)纯化粗产物,得到无色油状物,其含有29和28。通过Kugelrohr蒸馏进一步纯化该混合物,得到无色油状化合物29(0.44 g,19%)。GCMS:(222,M+);1H NMR(400 MHz,CDCl3)δH:(2︰1的酮和烯醇互变异构体的混合物)烯醇:11.37 (1H,s),7.36-7.28(2H,m),7.07-6.88(3H,m),4.21 (2H,q,J=7.1),1.99(3H,s),1.18(3H,t,J=7.1);酮:7.36-7.28(2H,m),7.07-6.88(3H,m),5.09(1H,s),4.30(2H,q,J=7.1),2.39(3H,s),1.30(3H,t,J=7.1)。
1.1.22 2-[2,6-二氯-3-(三氟甲基)苯氧基]-3-氧代丁酸乙酯(31)的合成
在0 ℃,向氢化钠(0.12 g,3.11 mmol,矿物油中的60%分散体)的四氢呋喃(6.0 mL)悬浮液中加入2,6-二氯-3-(三氟甲基)苯酚(0.72 g,3.11 mmol)。搅拌反应30 min,加入2-氯-3-氧代-丁酸乙酯28(0.52 mL,3.74 mmol)。将混合物加热至室温并搅拌1 h。将反应混合物加热回流18 h,然后冷却至室温。用Et2O (10 mL)稀释,然后用H2O(10 mL)洗涤。水层用Et2O (2×10 mL)萃取,合并有机萃取液并用NaOH水溶液(5.0 mL,2.0 M)洗涤,然后用MgSO4干燥,过滤并真空浓缩。柱色谱法纯化残留物,得到无色油状化合物31(436 mg,39%),其中含有微量的化合物28,无需进一步纯化即可使用。GCMS:(359,M+);1H NMR (400 MHz,CDCl3)δH:(7.5︰1酮和烯醇互变异构体的混合物)酮:7.50-7.33(2H,m),5.06(1H,s),4.27(2H,q,J=7.1),2.54(3H,s),1.28(3H,t,J=7.1);烯醇:11.23-11.19(1H,m),7.50-7.33(2H,m),4.14-4.04(2H,m),2.19(3H,s),1.03(3H,t,J=7.2)。
1.1.23 一般程序4——2-芳氧基--酮酯
将NaOH(1.1当量)的水溶液(0.1 mL/mmol)加入2,6-二氯苯酚(1.0当量)的甲苯(2.0 mL/mmol)溶剂中。将混合物加热至80 ℃下反应1 h,然后冷却至室温并真空浓缩,得到相应的苯酚钠。将残留物溶于DMF(2.0 mL/mmol)中,点滴加入相关的2-氯-酮酯(1.0~1.2当量)。在室温下搅拌反应液3 h。用Et2O (10 mL/mmol)稀释,用H2O(2×10 mL/mmol)洗涤。合并有机萃取物,用MgSO4干燥,过滤并真空浓缩。快速柱色谱或重结晶纯化粗产物,得到相应的2-芳氧基--酮酯。
1.1.24 2-(2,6-二氯苯氧基)-3-氧代丁酸乙酯(33)的合成
在一般方法中加入2,6-二氯苯酚(20.0 g,123 mmol)和2-氯-3-氧代丁酸乙酯28(20.4 mL,147 mmol)制备产物。用乙酸乙酯重结晶纯化粗产物,得到白色固体化合物33(27.3 g,76%)。GCMS:(290,M+);1H NMR(400 MHz,CDCl3)δH:(4.4︰1的酮:烯醇互变异构体的混合物)酮:7.32(2H,d,J=8.1),7.08-7.02(1H,m),5.01(1H,s),4.27(2H,q,J=7.2),2.54(3H,s),1.29(3H,t,J=7.2);烯醇:11.21(1H,s),7.26(2H,d,J=8.2),6.95(1H,d,J=8.1),4.13(2H,q,J=7.1),2.14(3H,s),1.08(3H,t,J=7.1)。
1.1.25 2-(2,6-二氯苯氧基)-3-氧代-戊酸乙酯(34)的合成
在一般方法4中加入2,6-二氯苯酚(2.8 g,17.2 mmol)和2-氯-3-氧代戊酸乙酯(3.1 g,17.2 mmol)制备产物。快速柱色谱纯化(1︰19的乙酸乙酯和异己烷)粗产物,得到无色油状物34(3.8 g,72%)。GCMS:(304,M+);1H NMR(400 MHz,CDCl3)δH:(11︰1的酮和烯醇互变异构体的混合物)酮:7.35-7.29(2H,m),7.08-7.01(1H,m),5.07(1H,s),4.26(2H,q,J=7.1),3.08(1H,dq,J=19.1和7.2),2.85(1H,dq,J=19.1和7.1),1.28(3H,t,J=7.1)1.16(3H,t,J=7.2);烯醇:11.29(1H,s),7.25(2H,d,J=8.1),6.93(1H,m,J=7.9),4.14(2H,q,J=7.1),2.53(2H,q,J=7.7),1.18-1.13(3H,m),1.08(3H,t,J=7.1)。
1.1.26 一般方法5——2芳氧基--酮酯与3-氨基三唑或3-氨基吡唑的缩合
将2-芳氧基--酮酯(1.0~2.0当量)加入相应的3-氨基唑(1.0当量)的乙酸(1.0 mL/mmol)溶液中,加热回流反应6 h,然后冷却至室温。将所得沉淀过滤,用H2O(1.0 mL/mmol)洗涤,真空干燥,得到相应的唑并嘧啶。
1.1.27 5-甲基-6-苯氧基-[1,2,4]三唑并[1,5-a]嘧啶-7-醇(35)的合成
在一般方法5中加入酮酯29(0.4 g,1.79 mmol)和3-氨基-1,2,4-三唑15(0.15 g,1.79 mmol)制备产物,得到白色固体35(0.27 g,63%)。LRMS:(243,M+H+);1H NMR(400 MHz,DMSO-d6)δH:13.40(1H,br.s),8.27(1H,s),7.34-7.23(2H,m),7.08-6.95(3H,m),2.25(3H,s);13C NMR(125MHz,DMSO-d 6)δC:157.4,152.6,152.1,149.2,145.0,129.6,125.7,122.1,114.7,14.4;mp:282~292 ℃。
1.1.28 6-(2,6-二氯苯氧基)-5-甲基-[1,2,4]三唑并[1,5-a]嘧啶-7-醇(36)
在一般程序5中加入酮酯33(1.0 g,3.43 mmol)和3-氨基-1,2,4-三唑15(0.29 g,3.43 mmol)制备产物。用CH2Cl2(5.0 mL)研磨进一步纯化产物,得到白色固体物36(0.56 g,54%)。LRMS:(311,M+H+);1H NMR(400 MHz,DMSO-d6)δH:13.54(1H,br.s),8.28(1H,s),7.49-7.43(2H,m),7.17(1H,t,J=8.1),2.47(3H,s);mp:289~296 ℃。
1.1.29 6-[2,6-二氯-3-(三氟甲基)苯氧基]-5-甲基-[1,2,4]三唑并[1,5-a]嘧啶-7-醇(37)的合成
在一般程序5中加入酮酯31(0.20 g,0.56 mmol)和3-氨基-1,2,4-三唑15(47 mg,0.56 mmol)进行制备产物。用热MeCN(3×5.0 mL)研磨进一步纯化产物,得到浅黄色固体37(34 mg,16%)。LRMS:(379,M+H+);1H NMR(400 MHz,DMSO-d6)δH:8.28(1H,s),7.64-7.72(2H,m),7.49(1H,s),2.50(3H,s);mp:231~237 ℃。
1.1.30 6-[2,6-二氯-3-(三氟甲基)苯氧基]-5-甲基-2-(三氟甲基)-[1,2,4]三唑并[1,5-a]嘧啶-7醇(38)
在一般程序5中加入酮酯31(1.00 g,2.78 mmol)和3-(三氟甲基-1-1,2,4-三唑-5-胺(0.42 g,2.78 mmol)制备产物。用快速柱色谱法(1︰19的MeOH和CH2Cl2)进一步纯化产物,得到为白色固体38(90 mg,7%)。LRMS:(447,M+H+);1H NMR(400 MHz,DMSO-d6)δH:7.75-7.65(2H,m),2.54(3H,s);mp:287~293 ℃。
1.1.31 6-[2,6-二氯-3-(三氟甲基)苯氧基]-2-异丙基-5-甲基-[1,2,4]三唑并[1,5-a]嘧啶-7-醇的合成(39)
在一般程序5中加入酮酯31(1.00 g,1.17 mmol)和5-异丙基-4-1,2,4-三唑-3-胺(0.30 g,2.38 mmol)制备产物。用快速柱色谱法(1︰19的MeOH和CH2Cl2)进一步纯化粗产物,得到黄色固体39(0.70 g,70%)。LRMS:(421,M+H+);1H NMR(400 MHz,DMSO-d6) δH:7.71-7.61(2H,m),3.02(6H,spt,J= 6.9),2.47(3H,s),1.26(6H,d,J=6.8);mp:128~142 ℃。
1.1.32 6-(2,6-二氯苯氧基)-5-乙基-[1,2,4]三唑并[1,5-a]嘧啶-7-醇(40)的合成
在一般方法5中加入酮酯34(1.2 g,3.9 mmol)和3-氨基-1,2,4-三唑15(0.27 g,3.25 mmol)制备产物。用快速柱色谱法(1︰9的MeOH和CH2Cl2)进一步纯化粗产物,得到棕色固体40(0.36 g,35%)。LRMS:(325,M+H+);1H NMR(400 MHz,DMSO-d6) δH:13.57(1H,br.s),8.32(1H,s),7.49-7.41(2H,m),7.21-7.12(1H,m),2.84 (2H,q,J=7.5),1.29(3H,t,J=7.6);mp:>300 ℃。
1.1.33 6-(2,6-二氯苯氧基)-5-乙基-2-(三氟甲基)-[1,2,4]三唑并[1,5-a]嘧啶-7-醇(41)的合成
在一般方法5中加入酮酯34(1.2 g,3.9 mmol)和5-(三氟甲基)-4-1,2,4-三唑-3-胺(0.29 g,1.95 mmol)制备产物。用快速柱色谱法(1︰9的MeOH和CH2Cl2)进一步纯化粗产物,得到黄色固体41(130 mg,17%)。 LRMS:(393,M+H+);1H NMR(400 MHz,DMSO-d6) δH:7.50-7.45(2H,m),7.19(1H,t,J=8.1),2.88(2H,q,J=7.6),1.31(3H,t ,J= 7.6);mp:193~202 ℃。
1.1.34 6-(2,6-二氯苯氧基)-5-乙基-2-异丙基-[1,2,4]三唑并[1,5-a]嘧啶-7-醇(42)的合成
在一般方法5中加入酮酯34(1.20 g,3.9 mmol)和5-异丙基-4-1,2,4-三唑-3-胺(0.41 g,3.24 mmol)制备产物。用快速柱色谱法(1︰9的MeOH和CH2Cl2)进一步纯化粗产物,得到棕色固体42(431 mg,36%)。 LRMS:(367,M+H+);1H NMR(400 MHz,DMSO-d6) δH:13.28(1H,br.s),7.44(2H,d,J=8.2),7.15(1H,t,J=8.1),3.03(1H,spt ,J=6.9),2.80(2H,q,J=7.6),1.31-1.23(9H,m);mp:109~112 ℃。
1.1.35 6-(2,6-二氯苯氧基)-5-甲基-2-吗啉代-[1,2,4]三唑并[1,5-a]嘧啶-7-醇(43)的合成
在一般方法5中加入酮酯29(0.86 g,2.96 mmol)和3-吗啉代-1-1,2,4-三唑-5-胺(0.50 g,2.96 mmol)制备产物,得到浅黄色固体43(0.71 g,61%)。LRMS:(396,M+H+);1H NMR(400 MHz,DMSO-d6)δH:13.18-13.14(1H,m),7.44(2H,d,J= 8.1),7.15(1H,t,J=8.1),3.67(4H,t,J=4.6),3.42-3.28(4H,m),2.39(3H,s); mp:>300 ℃
1.1.36 6-(2,6-二氯苯氧基)-2-(二甲基氨基)-5-甲基-[1,2,4]三唑并[1,5-a]嘧啶-7-醇(44)的合成
在一般方法5中加入酮酯33(1.14 g,3.93 mmol)和3,3-二甲基-1-1,2,4-三唑-3,5-二胺(0.50 g,3.93 mmol)制备产物,得到浅黄色固体44(0.81 g,58%)。LRMS:(354,M+H+);1H NMR(400 MHz,DMSO-d6)δH:13.20-12.93(1H,m),7.44(2H,d,J=8.1),7.15(1H,t,J=8.1),2.94(6H,s),2.37(3H,s):mp:>300 ℃。
1.1.37 6-(2,6-二氯苯氧基)-5-甲基-2-(三氟甲基)-[1,2,4]三唑并[1,5-a]嘧啶-7-醇(45)的合成
在一般方法5中加入酮酯33(0.96 g,3.29 mmol)和3-(三氟甲基)-1-1,2,4-三唑-5-胺(0.50 g,3.29 mmol)制备产物,冷却时不形成沉淀,真空浓缩。把残留物悬浮于CH2Cl2(5.0 mL)中,形成沉淀物。过滤,将滤液真空干燥,得到浅黄色固体45(0.37 g,29%)。LRMS:(379,M+H+);1H NMR(400 MHz,DMSO-d6) δH:12.72(1H,br.s.),7.50-7.45(3H,m),7.19(1H,t,J=8.1),2.51(3H,s);mp:228~229 ℃。
1.1.38 2-环丙基-6-(2,6-二氯苯氧基)-5-甲基-[1,2,4]三唑并[1,5-a]嘧啶-7-醇(46)的合成
在一般方法5中加入酮酯33(1.17 g,4.03 mmol)和3-环丙基-1-1,2,4-三唑-5-胺(0.50 g,4.03 mmol)制备产物。混合物真空浓缩,产生沉淀。将混合物过滤,用AcOH(2.0 mL)和CH2Cl2(2.0 mL)洗涤,然后真空干燥,得到浅黄色固体46(0.81 g,57%)。LRMS:(351,M+H+);1H NMR(400 MHz,DMSO-d6) δH:13.41-12.98(1H,m),7.45(2H,d,J= 8.2),7.16(1H,t,J=8.1),2.42(3H,s),2.08-1.99(1H,m),1.05-0.97 (2H,m),0.93-0.85(2H,m)。mp:279~288 ℃。
1.1.39 6-(2,6-二氯苯氧基)-2-(甲氧基甲基)-5-甲基-[1,2,4]三唑并[1,5-a]嘧啶-7-醇(47)的合成
在一般方法5中加入酮酯33(1.14 g,3.90 mmol)和3-(甲氧基甲基)-1-1,2,4-三唑-5-胺(0.50 g,3.90 mmol)制备产物,得到47白色固体(0.69 g,49%)。LRMS:(355,M+H+);1H NMR(400 MHz,DMSO-d6)δH:13.79-13.26(1H,m),7.46(2H,d,J=8.2),7.21-7.13 (1H,m),4.47(3H,s),3.32 (3H,s),2.47(3H,s);mp:293~298 ℃。
1.1.40 6-(2,6-二氯苯氧基)-2-乙基硫烷基-5-甲基-[1,2,4]三唑并[1,5-a]嘧啶-7-醇(48)的合成
在一般方法5中加入酮酯33(1.01 g,3.47 mmol)和3-乙基硫烷基-1-1,2,4-三唑-5-胺(0.50 g,3.47 mmol)制备产物,得到浅黄色固体48(0.61 g,48%)。LRMS:(371,M+H+);1H NMR(400 MHz,DMSO-d6)δH:13.44(1H,br. s),7.46(2H,d,J=8.1),7.17(1H,t,J=8.1),3.12(2H,q,J=7.3),2.44(3H,s),1.34(3H,t,J=7.3);mp:297~98 ℃。
1.1.41 6-(2,6-二氯苯氧基)-2-甲氧基-5-甲基-[1,2,4]三唑并[1,5-a]嘧啶-7-醇(49)的合成
在一般方法5中加入酮酯33(1.02 g,3.51 mmol)和3-甲氧基-1H-1,2,4-三唑-5-胺(0.40 g,3.51 mmol)制备产物,得到白色固体49( 0.49 g,41%)。LCMS:(341,M+H+);1H NMR(400 MHz,DMSO-d6)δH:13.50-13.22(1H,m),7.46(2H,d,J=8.1),7.17(1H,t,J=8.1),3.92(3H,s),2.41(3H,s);mp:286~290 ℃。
1.1.42 6-(2,6-二氯苯氧基)-2-异丙基-5-甲基-[1,2,4]三唑并[1,5-a]嘧啶-7-醇(50)的合成
在一般方法5中加入酮酯33(0.92 g,3.17 mmol)和3-异丙基-1-1,2,4-三唑-5-胺(0.40 g,3.17 mmol)制备产物,得到白色固体(0.30 g,27%)。LRMS:(353,M+H+);1H NMR(400 MHz,DMSO-d6)δH:13.30-12.85 (1H,m),7.45(2H,d,J= 8.1),7.16(1H,t,J=8.1),3.02(1H,spt,J= 6.9),2.43(3H,s),1.26(6H,d,J=7.0);mp:275~282 ℃。
1.1.43 6-(2,6-二氯苯氧基)-2,5-二甲基-[1,2,4]三唑并[1,5-a]嘧啶-7-醇(51)的合成
在一般方法5中加入酮酯33(0.30 g,1.03 mmol)和3-甲基-1-1,2,4-三唑-5-胺(0.10 g,1.03 mmol)制备产物,得到白色固体51(60 mg,18%)。LRMS:(325,M+H+);1H NMR(400 MHz,DMSO-d6)δH:7.45(2H,d,J=7.3),7.17-7.14(1H,m),2.43(3H,s),2.34(3H,s);mp:>300 ℃。
1.1.44 6-(2,6-二氯苯氧基)-5-甲基-2-(1-甲基环丙基)吡唑并[1,5-a]嘧啶-7-醇(52)的合成
在一般方法5中加入酮酯33(0.84 g,2.88 mmol)和3-(1-甲基环丙基)-1-吡唑-5-胺(0.50 g,2.88 mmol)制备产物,得到白色固体52(0.39 g,37%)。LRMS:(364,M+H+);1H NMR(400 MHz,DMSO-d6)δH:12.29(1H,br.s.),7.43(2H,d,J= 8.1),7.14(1H,t,J=8.1),5.84(1H,s),2.42(3H,s),1.39(3H,s),0.96(2H,br.s.),0.77(2H,br.s.);mp:>300 ℃。
1.1.45 6-(2,6-二氯苯氧基)-7-羟基-5-甲基-吡唑并[1,5-a]嘧啶-2-甲腈(53)的合成
在一般方法5中加入酮酯33(0.98 g,3.38 mmol)和5-氨基-1-吡唑-3-甲腈(0.37 g,3.38 mmol)制备产物,得到桃色固体53(0.85g,75 %)。LRMS:(355,M+H+);1H NMR(400 MHz,DMSO-d6)δH:13.90-13.42 (1H,m),8.39(1H,s),7.46(2H,d,J= 8.1),7.18(1H,t,J=8.1),2.50(3H,s);mp:>300 ℃。
1.1.46 3-溴-6-(2,6-二氯苯氧基)-5-甲基吡唑并[1,5-a]嘧啶-7-醇(54)的合成
在一般方法5中加入酮酯33(0.42 g,1.43 mmol)和4-溴-1-吡唑-5-胺(0.23 g,1.43 mmol)制备产物,得到浅紫色固体54(0.25 g,47%)。LRMS:(390,M+H+);1H NMR(400 MHz,DMSO-d6)δH:12.66(1H,br.s.),8.04(1H,s),7.49(2H,d,J=8.1),7.24-7.16(1H,m),2.55(3H,s);mp:281~283 ℃。
1.1.47 6-(2,6-二氯苯氧基)-5-甲基-3-硝基吡唑并[1,5-a]嘧啶-7-醇(55)的合成
在一般方法5中加入酮酯33(1.26 g,4.32 mmol)和4-硝基-1-吡唑-5-胺(0.55 g,4.32 mmol)制备产物,得到白色固体55(1.09 g,71%)。LRMS:(355,M+H+);1H NMR(400 MHz,DMSO-d6)δH:13.40-12.58 (1H,m),8.65(1H,s),7.52-7.43(2H,m),7.20(1H,t,J=8.1),2.61(3H,s);mp:292~297 ℃。
1.1.48 6-(2,6-二氯苯氧基)-2-乙氧基-7-羟基-5-甲基-吡唑并[1,5-a]嘧啶-3-甲腈(56)的合成
在一般方法5中加入酮酯33(0.65 g,2.24 mmol)和5-氨基-3-乙氧基-1-吡唑-4-甲腈(0.34 g,2.24 mmol)制备产物,得到白色固体56(0.52 g,61%)。LRMS:(379,M+H+);1H NMR(400 MHz,DMSO-d6)δH:13.67-13.28 (1H,m),7.48-7.42(2H,m),7.17(1H,t,J=8.1),4.32(2H,q,J=7.0),2.44(3H,s),1.35(3H,J=7.0);mp:271~280 ℃。
1.1.49 6-(2,6-二氯苯氧基)-5-甲基-吡唑并[1,5-a]嘧啶-7-醇的合成(57)
在一般方法5中加入酮酯33(1.40 g,4.82 mmol)和1-吡唑-5-胺(0.40 g,4.82 mmol)制备产物,得到白色固体57(1.02 g,69%)。LRMS:(310,M+H+);1H NMR(400 MHz,DMSO-d6)δH:12.52(1H,br.s.),7.86(1H,d,J=2.0),7.47-7.42(2H,m),7.15(1H,t,J=8.1),6.12(1H,d,J=2.0),2.46(3H,s);mp:>300 ℃。
1.1.50 6-(2,6-二氯苯氧基)-7-羟基-2,5-二甲基-吡唑并[1,5-a]嘧啶-3-甲腈(58)的合成
在一般方法5中加入酮酯33(0.72 g,2.47 mmol)和5-氨基-3-甲基-1-吡唑-4-甲腈(0.30 g,2.47 mmol)制备产物,得到白色固体58(0.61 g,70%)。LRMS:(349,M+H+);1H NMR(400 MHz,DMSO-d6)δH:13.72-13.29(1H,m),7.46(2H,d,J=8.2),7.21-7.12 (1H,m),2.47(3H,s),2.35(3H,s);mp:>300 ℃。
1.1.51 6-[2,6-二氯-3-(三氟甲基)苯氧基]-7-羟基-5-甲基-吡唑并[1,5-a]嘧啶-3-甲腈(59)的合成
在一般方法5中加入酮酯31(0.20 g,0.56 mmol)和5-氨基-1-吡唑-4-丁腈(60 mg,0.56 mmol)制备产物,得到棕色固体59(63 mg,28%)。LRMS:(403,M+H+);1H NMR(400MHz,DMSO-d6)δH:14.14-13.30 (1H,m),8.41(1H,s),7.76-7.62(2H,m),2.53(3H,s)。
1.1.52 6-(2,6-二氯苯氧基)-7-羟基-5-甲基-吡唑并[1,5-a]嘧啶-3-甲腈(60)的合成
在一般方法5中加入酮酯33(2.69 g,9.25 mmol)和5-氨基-1-吡唑-4-丁腈(1.00 g,9.25 mmol)制备产物,得到棕色固体状60(2.14 g,69%)。LRMS(335,M+H+);1H NMR(400 MHz,DMSO-d6)δH:13.60 (1H,br.s),8.39(1H,s),7.46(1H,d,J=8.0),7.18(1H,t,J=8.0),2.50(3H,s)。
1.2 X射线晶体学
将化合物18、36和60的单晶样品安装在Paratone N(Hampton Research,Aliso Viejo,CA)上,进行CuK衍射(Agilent,Oxford,UK),于173 ℃ Excalibur PX Ultra衍射仪收集数据。用CrysAlis Pro(Agilent)处理数据,用abspack module进行吸收校正。用SIR92解析结构,用CRYSTALS对结构进行精修。
1.3 除草活性的生物测定
根据以下方法评价了化合物对龙葵(,SN)、反枝苋(,AR)、大狗尾草(,SF)、黑麦草(,LP)、稗草(,EC)和裂叶牵牛(,IP)的芽后除草活性。将这些杂草的种子播种在盆栽试验的标准土壤中。控制温室(在24/16 ℃,白天/晚上,14 h光照,65%湿度)条件下,种子萌发后培育8 d,用含有0.5%吐温20(聚氧乙烯脱水山梨醇单月桂酸酯,CAS RN 9005-64-5)的丙酮-H2O(50︰50)活性成分的丙酮-水(50︰50)溶液喷雾。在以上条件下继续培养,且每天浇水2次。13 d后,目视评估测试化合物对植物的毒性(100=对植物的造成完全损害;0=对植物没有损害)。施用浓度为有效成分1 000 g/hm2,施用量为1 000 L/hm2。
1.4 测定氨基三唑的产生
1.4.1 最初氨基三唑测定试验
根据以下方法评估植物中氨基三唑化合物15的产生。反苋枝长至3叶期时,分别用化合物37、61、62和63以有效成分500 g/hm2、200 L/hm2喷雾。氨基三唑化合物以125 g/hm2施用。每个喷雾处理结束时用10 mL乙腈-水(80︰20)清洗喷雾器。用10 mL乙腈-水(80:20)洗涤叶子表面残留物。然后在施药后24、96 h用2×3.0 mL乙腈-水(80︰20)萃取叶子,取100 μL萃取液稀释至1.0 mL,用于LCMS分析。检测仪器为Thermo TSQ Quantum Discovery MS(装有Waters Acquity sample和solventmanager)和Obelisc R柱(150 mm×2.1 mm),用甲酸铵的乙腈-水[洗脱剂A:5.0 mM的甲酸铵水溶液;洗脱剂B:5.0 mM甲酸铵的乙腈-水(95︰5)溶液;方法:T0-0.5 min,2%A,T3-3.1min80%A,T3.2-4 min2%A]溶液进行梯度洗脱,流速:0.5 mL/min。Amitrole 15的保留时间为1.93 min。通过与基质匹配标准比较确定检出率。
1.4.2 进一步氨基三唑测定试验
反枝苋在2叶期时,分别对其进行氨基三唑化合物15和化合物2、61、64和65溶液手动喷雾,剂量为200 μg/mL,喷雾体积为110 μL。用4.0 mL乙腈-水(80︰20)溶液冲洗叶子表面的残留物。然后在施用后24、48 h,用4.0 mL乙腈-水(80︰20)萃取叶子,取1.0 mL萃取液进行LCMS分析。检测仪器为Thermo TSQ Quantum Discovery MS(装有Waters Acquity sample和solventmanager)和Obelisc R柱(150 mm×2.1 mm),用甲酸铵的乙腈-水[洗脱剂A:5.0 mM的甲酸铵水溶液;洗脱剂B:5.0 mM甲酸铵的乙腈-水(95︰5)溶液;方法:T0-0.5 min,2%A,T3-3.1min80%A,T3.2-4 min2%A]溶液进行梯度洗脱,流速:0.5 mL/min。Amitrole 15的保留时间为1.93 min。通过与基质匹配标准比较确定检出率。
1.5 用过度表达IspD和番茄红素-环化酶的植物进行研究
1.5.1 载体构建
进行IspD(AT2G02500,NCBI ref:NP_565286)和番茄红素-环化酶(AT3G10230,NCBI ref:NP_187634)序列密码子优化以烟草表达,并用来自Genewiz(South Plainfield,NJ)的烟草花叶病毒(EMBL:TOTMV6)的5′前导序列合成。合成的片段在5′末端具有Xhol酶切位点,在3'末端具有Kpnl酶切位点。将合成的基因克隆到卡那霉素抗性的植物二元载体中,作为35S启动子后面和NOS终止子前的Xhol/Kpnl片段。
1.5.2 植物转化
如前所述烟草(var. Samsung)的过度表达被终止了。用含有用于过度表达IspD或番茄红素-环化酶(LYC)的植物双元载体的根癌农杆菌菌株LBA4404来转换叶碟。通过单一转化产生了卡那霉素抗性烟草,并用PCR确定了转基因的存在。
1.5.3 除草活性的生物测定
大约在转基因植物3叶期,将这些植物进行盆栽,并在标准温度条件下培养一周。喷雾设备距离植物30 cm处喷雾,喷雾量200 L/hm2。对每个转基因株系的一个克隆植物进行喷雾。对过度表达IspD的转基因系,氨基三唑15以60 g/hm2和120 g/hm2的剂量施用;化合物37、61、62和63的剂量为500 g/hm2,化合物45、64和65的剂量为2 000 g/hm2。
对于过度表达番茄红素-环化酶的转基因系,氨基三唑15的用量为60 g/hm2和120 g/hm2,化合物61的为500 g/hm2,化合物62的为300 g/hm2,化合物64和65的施用量为2 000 g/hm2。
2 结果与讨论
2.1 化学合成
新型芳氧基系列4的化合物可分为2个不同的化学类别(方案1)。羟基氯代偶氮嘧啶(hydroxychloro azolopyrimidine)的合成[图3,(1)]将需要最初的3-氨基唑5与2-芳氧基丙二酸酯6的缩合,然后如前所述进行2次进一步的化学转化。设想3-氨基唑5与芳氧基-酮酯7直接缩合将得到在嘧啶基上含有烷基的唑并嘧啶4[图3,(2)]。
芳氧基系列的合成开始于制备各种2-芳氧基丙二酸酯(图4)。苯酚系列8和氯代丙二酸二乙酯9在碱性条件下的反应,得到中等收率的芳氧基丙二酸酯10。类似地,许多容易获得的苯酚可以良好至高的产率转化为相应的丙二酸酯11-14。
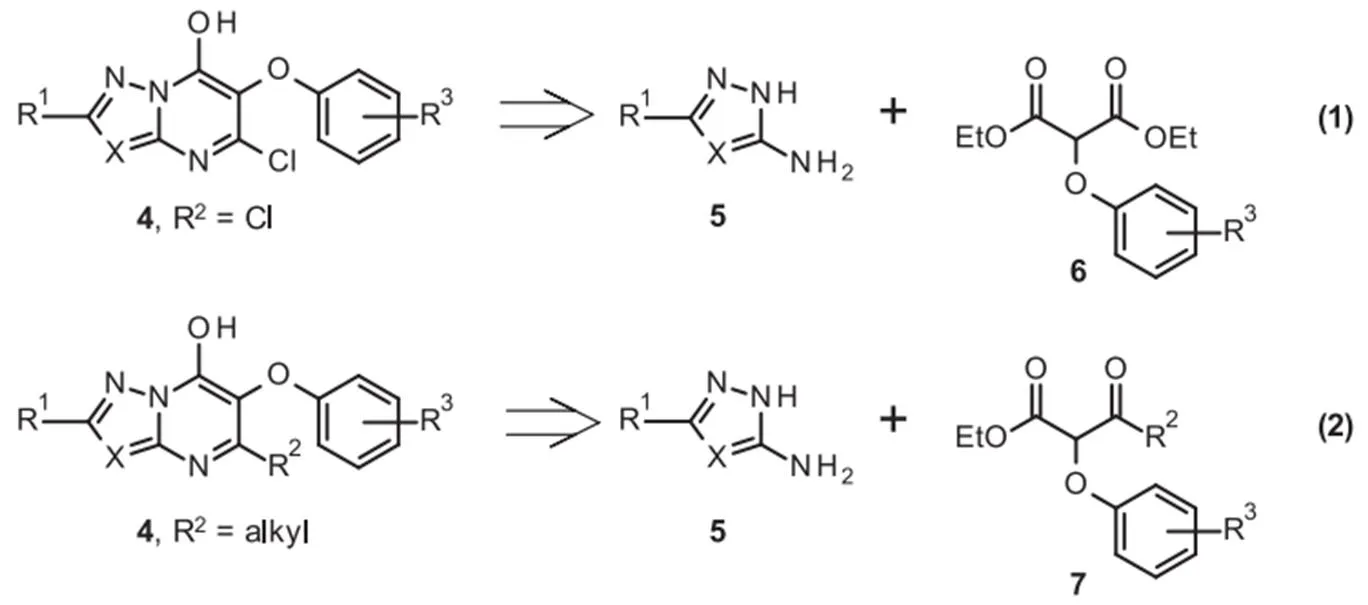
图3 芳氧基系列4的逆合成

图4 2-芳氧基丙二酸酯的制备
丙二酸酯10与3-氨基-1,2,4-三唑(amitrole)15缩合,得到相应的嘧啶-5,7-二醇16,其通过两步以良好的收率直接转化为二氯化物17(图5)。用氢氧化钠部分水解17,得到异构体的混合物,用制备型HPLC分离,以61%的产率得到所需的7-羟基三唑并嘧啶18以及10%的5-羟基三唑并嘧啶19。
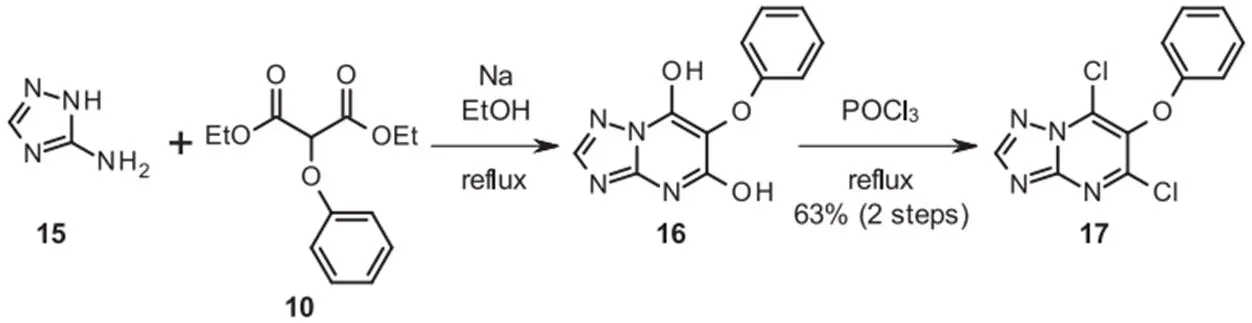
图5 二氯芳氧基嘧啶18的合成
主要的异构体18的结构经X衍射晶体学分析得到(图6)。发现三唑并嘧啶18晶体中存在N-H互变异构体结构。
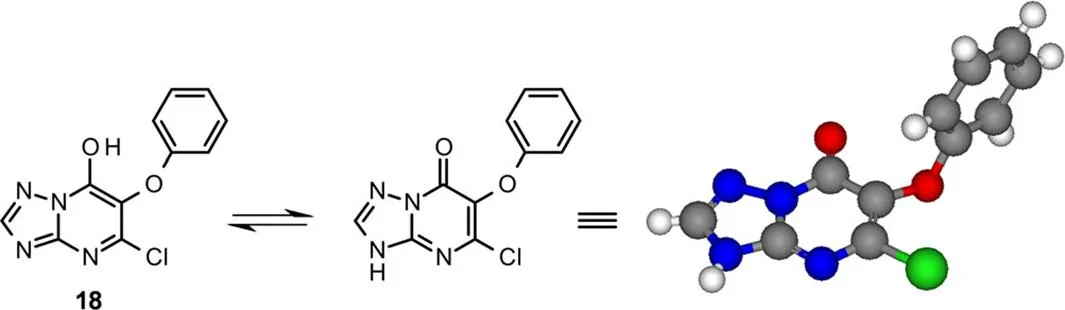
图6 化合物18的X-衍射晶体结构
令人高兴的是,冷凝和氯化化学可应用于合成其他的丙二酸酯系列(表1)。通过2步,以中等(表1,编号3和4)到良好的产率合成二氯嘧啶20至23(表1,编号1和2)。
在相同的基本条件下,合成了所需的羟基三唑并嘧啶24至27。在所有情况下,7-羟基-三唑并嘧啶是主要的区域异构体(表2,编号1至4)。
接下来,制备了一系列2-芳氧基-酮酯(图7)。由化合物8原位合成苯酚钠,然后与2-氯-3-氧代丁酸乙酯28反应,以19%中等分离产率得到相应的2-芳氧基-酮酯29。
类似地苯酚化合物30生成了醚31,产率提高到了39%。对反应条件进行筛选发现在与-氯代-酮酯反应之前,用氢氧化钠形成和分离化合物32的苯酚钠,得到相应的醚33和34,且产率提高了(图8)。

表1 芳氧基丙二酸酯经2步反应生成二氯化物
注:a2步反应的分离率。
在酸性条件下,醚29与氨基三唑15缩合得到相应的三唑并嘧啶35,产率63%(表3,编号1)。令人高兴的是,这些反应条件可以应用于广泛的3-氨基三唑(表3,编号1至17)和3-氨基吡唑(表3,编号18至26)的反应,以合成多种6-芳氧基-7-羟基唑并嘧啶。大多数6-芳氧基-7-羟基唑并嘧啶由醚化合物33合成,尽管醚34(表3,编号6至8)和31(表3,编号3-5和25)也可成功地反应。
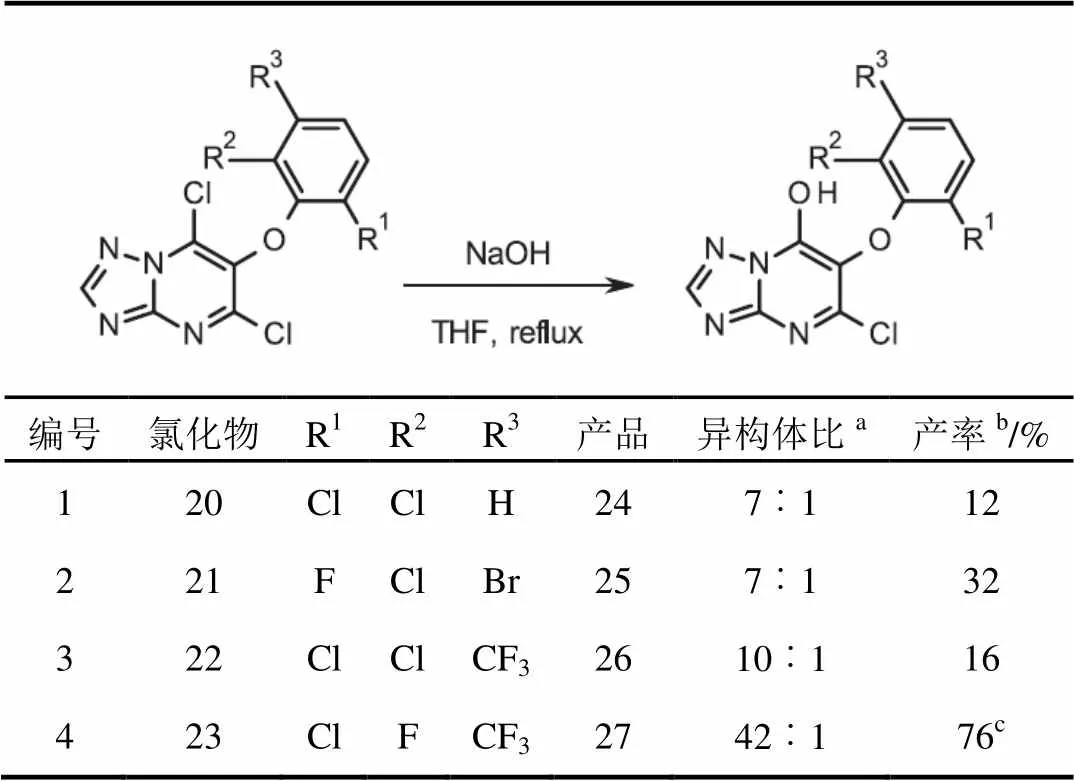
表2 羟基三唑并嘧啶的形成
注:a对粗反应混合物进行1HNMR分析所得;b7-羟基异构体分离产率;c没有进一步纯化,产品为4︰1异构体的混合物。

图7 用NaH合成2-芳氧基-β酮酯29和31
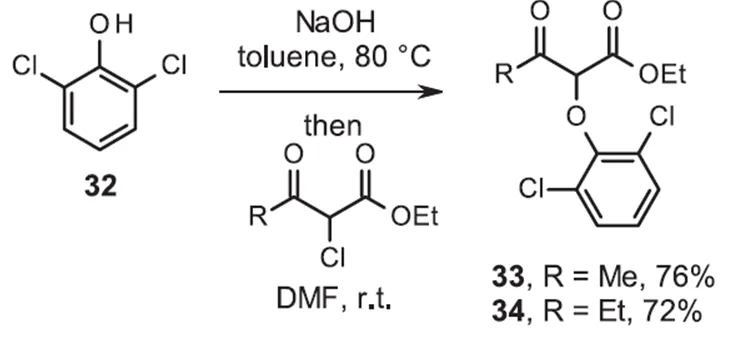
图8 用NaOH合成2-芳氧基β-酮酯
其中每种化合物都可被分离为单个区域异构体。对三唑衍生物36(用DMS8O-MeCN重结晶)和吡唑衍生物60(用H2O-MeCN中重结晶)进行X射线晶体学分析发现二者都为N-H的互变异构体,证实了缩合反应的区域选择性(图9)。
2.2 生物学特征
2.2.1 除草活性
以氨基三唑15和苄基化三唑并嘧啶2(图1)为标准,评估了所有新化合物对6种常见的杂草芽前和芽后的除草活性。试验发现芳氧基系列化合物没有显著的芽前的除草活性,大多数化合物的活性低于标准化合物(表4,编号3),所有化合物的活性都低于氨基三唑。然而,数个化合物相的除草活性与化合物2相似(表4,编号4和6)或高于其(表4,编号5)。
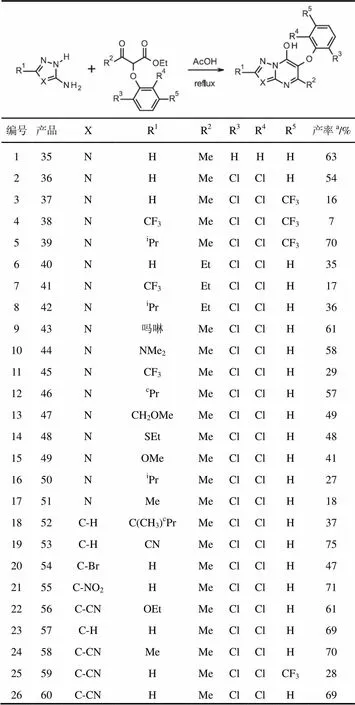
表3 5-烷基唑并嘧啶酮的形成
注:a分离产率。
在本研究中还测试了许多没有芳氧基或苄基取代基的化合物,例如已知为除草剂的简单的羟基-三唑并嘧啶61和相关的三唑并嘧啶62和63(表4,编号8至10),以及苄基化的三唑并嘧啶2的类似物(表4,编号11和12)。
2.2.2 除草症状
所有具有除草活性的化合物(表4,编号3至10),包括新颖芳氧基化合物(表4,编号3至7))引起了类似于引导化合物2的白化症状,表明它们都有同样的作用机制。有趣的是,这种漂白症状也与氨基三唑化合物 15所显示症状相似。显然,大部分的芳氧基系列化合物是氨基三唑经一步化学反应产生;尽管在样品的分析中没有检测到氨基三唑,但它仍然作为生测的一个标准。从其他氨基唑衍生而来的化合物的活性一般低于从氨基三唑衍生来的化合物。氨基三唑化合物由于不利的毒理特性,在欧盟和美国作为非选择性除草剂的非农用途受到了限制。这当然意味着在植物中能产生氨基三唑的任何新类别的除草剂将不会具有吸引力的商业前景。氨基三唑15的除草靶标未知。各种研究表明,此物质抑制植物酶八氢番茄红素去饱和酶、番茄红素环化酶、咪唑甘油-磷酸脱水酶和过氧化氢酶,但已排除抑制组氨酸生物合成的作用为其作用机制。另外,有人提出,抗禾草灵的瑞士黑麦草体内氨基三唑抑制禾草灵羟化酶,即细胞色素P450微粒体氧化酶;这解释了氨基三唑化合物和禾草灵对此种抗性虫增效试验中禾草灵代谢水平降低的原因。为了弄明白化合物的白化作用的缘由,挑选了一些化合物,包括芳氧基系列4的化合物,研究了它们在植物体内代谢和抗性情况。
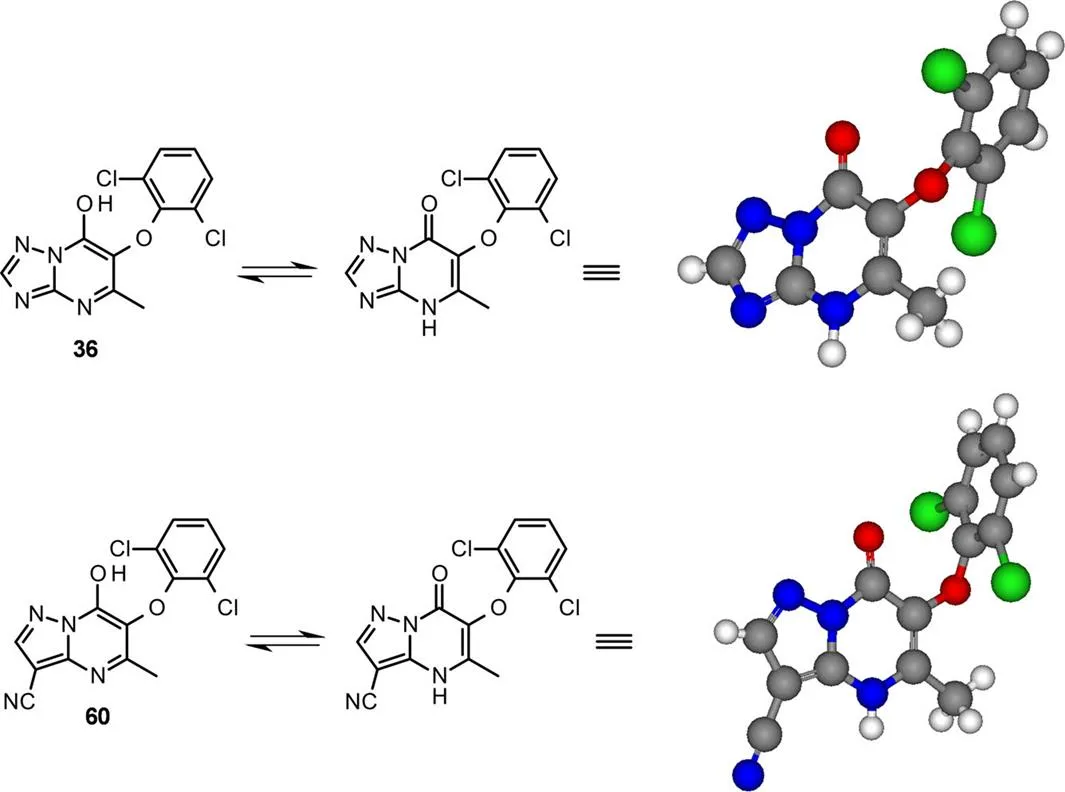
图9 混合物36和60的X-衍射晶体结构
2.3 植物代谢研究
为了确定三唑并嘧啶化合物白化作用是否由氨基三唑15所致,对反枝苋进行了叶萃取生测试验。化合物37、61、62和63以有效成分500 g/hm2施用,用LCMS测定了不同时间点叶提取物中的氨基三唑的量。
在处理后0、1、4 d测定叶提取物的结果表明所有处理中都产生了氨基三唑15。氨基三唑的产生是暂时的,在施用后1 d其量达到峰值,化合物61处理中氨基三唑15的量最大。4 d后,所有处理均表现出分生组织白化症状,与氨基三唑的相似,然而白化量与叶提取物中氨基三唑的水平无相关性。
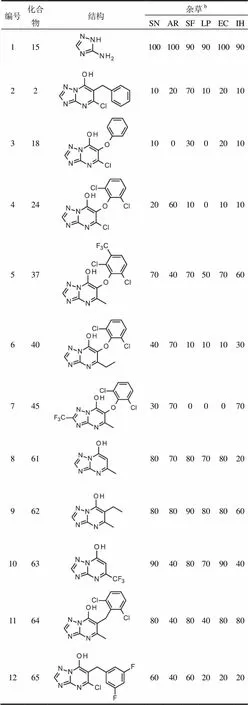
表4 芳氧基系列a化合物的选择性除草活性
注:a芽后除草效果;b杂草名称缩写见1.3节。
接着,以氨基三唑和化合物61为标准,测定了化合物2和其类似物64和65在植物中产生氨基三唑的情况。这些化合物的施用剂量比最初研究中的更低,检测到的氨基三唑的水平也低,但发现化合物2、64和65在反枝苋体中产生显着水平的氨基三唑。在此较低的浓度水平,处理中几乎没有白化症状发生。然而,所有处理都出现坏死,叶片缩水和卷曲的症状,这可能是由于产生亚致死剂量的氨基三唑,新生长点受损所致。这些试验结果与假设的情况一致,即假设观察到除草活性是由于植物体内生成氨基三唑所致。
2.4 作用机制的研究
2.4.1 植物过度表达IspD
如上所述培育了表达高水平IspD的烟草。测定了活性最好的化合物即芳氧基系列化合物37和45以及化合物61至63对基因修饰和野生型烟草的除草活性。
所有化合物对基因修饰与野生型烟草的除草活性几乎没有差异(表5,条目1至5),这表明这些化合物的除草活性不是因为抑制IspD所致。有趣的是,用一些化合物处理时观察到的除草活性表明了IspD水平升高的适合度代价(表5,条目1至3,5至7和9)。
2.4.2 植物过度表达番茄红素-环化酶
如上所述培育了高水平表达番茄红素-环化酶的烟草。测试了氨基三唑15和化合物61、62、64和65对基因修饰和野生型烟草的除草活性。
氨基三唑15(表6,编号1和2)对过度表达番茄红素-环化酶的烟草的活性要低于对野生型的。烟草过度表达番茄红素-环化酶后对60 g/hm2的的氨基三唑具有耐受性,而对120 g/hm2氨基三唑具有部分耐受性。氨基三唑施用量为120 g/hm2时,番茄红素-环化酶抗性植物的白化程度比野生型植物降低很多。这些结果表明,氨基三唑15的作用机制为抑制番茄红素-环化酶的活性。化合物61和62对番茄红素-环化酶过度表达的烟草的除草活性(表6,编号3和4)低于对野生型烟草,前者的白化程度也低于后者。这与植物代谢研究结果都明显表明化合物61和62的除草活性是由于它们在植物体内生成了氨基三唑。化合物64和65对野生型和番茄红素-环化酶抗性烟草具有弱的除草活性,这难以确定它们对其他植物的作用方式,尽管它们在反枝苋体内生成了氨基三唑,并且过度表达IspD的植物对化合物64没有耐受性。
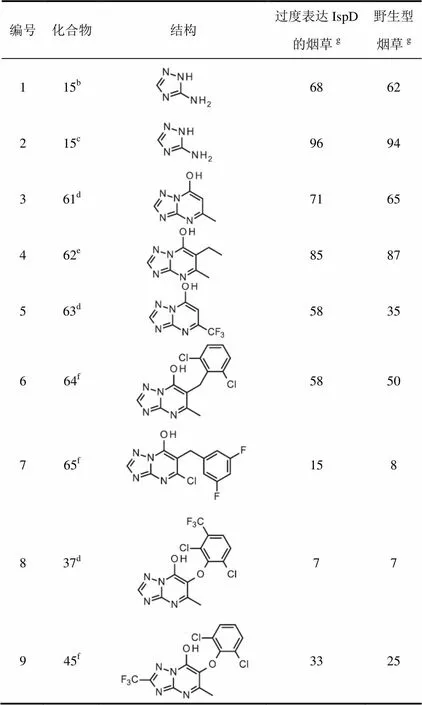
表5 化合物对野生型和过度表达IspD烟草除草活性a比较
注:a芽后防效(%,100=对整个植物有损害,0=对植物无损害);b剂量60 g/hm2;c剂量120 g/hm2;d剂量500 g/hm2;e剂量300g/hm2;f 2 000 g/hm2;g3个重复的平均值。
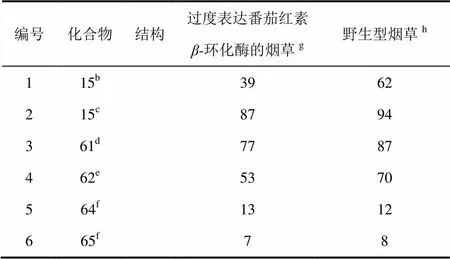
表6 化合物对野生型和过度表达番茄红素β-环化酶烟草的除草活性a比较
注:a芽后防效(%,100=对整个植物有损害,0=对植物无损害);b剂量60 g/hm2;c剂量120 g/hm2;d剂量500 g/hm2;e剂量300 g/hm2;f2 000 g/hm2;g4个重复的平均值;h4个重复的平均值。
3 结 论
制备了大量的芳族氧基系列4的羟基三唑并嘧啶和羟基吡唑并嘧啶化合物,并且测定了它们的除草活性和构效关系。只有羟基三唑并嘧啶亚类化合物具有显著的除草活性。此外,这一系列中只有少数几个化合物的活性比先导化合物2高,报道化合物2的除草活性作用机制为对植物中类异戊二烯生物合成的非甲羟戊酸途径的IspD有抑制作用。观察到这些除草剂与氨基三唑15有相似的白化症状,这意味着羟基三唑并嘧啶可能为氨基三唑的复杂前药。随后研究了化合物2、37和61在反枝苋体内代谢情况,结果证明确实如此。之后测定了氨基三唑15、化合物37、45和61-65对过度表达IspD的植物的除草活性,发现对IspD的抑制作用并不是化合物除草活性的作用机制。最后,测定了氨基三唑15和化合物61、62、64和65对过度表达番茄红素-环化酶植物的除草活性,结果为氨基三唑对基因修饰植物的活性显著低于对野生型的,表明其作用方式是抑制番茄红素-环化酶的活性。化合物61和62对过度表达番茄红素-环化酶植物的除草活性也降低了,这与植物代谢研究结果表明这些化合物之所以具有除草活性是因为它们在植物体内生成了氨基三唑。目前羟基三唑并嘧啶生成氨基三唑的生物化学机制尚不清楚,可能涉及氧化途径,如尿嘧啶的代谢。
总之,以上研究表明,商业产品氨基三唑的除草活性源于对植物中番茄红素-环化酶的抑制,本文所述的一些羟基三唑并嘧啶的除草活性是由于它们在植物中生成了氨基三唑。
10.16201/j.cnki.cn31-1827/tq.2017.03.02
TQ450
A
1009-6485(2017)03-0007-13
章乐天(1994—),男,汉族,在读硕士研究生。E-mail: zlt94zlt@163.com。
2017-05-10。
