戊乙奎醚光学异构体与毒蕈碱型受体亚型的分子对接研究
2017-10-10张丽娟白兰师健友何俊
张丽娟,白兰,师健友,何俊
(1.四川省医学科学院/四川省人民医院药学部,成都610072;2.四川大学华西医院肿瘤中心生物治疗实验室,成都 610041)
戊乙奎醚光学异构体与毒蕈碱型受体亚型的分子对接研究
张丽娟1*,白兰1,师健友1,何俊2#
(1.四川省医学科学院/四川省人民医院药学部,成都610072;2.四川大学华西医院肿瘤中心生物治疗实验室,成都 610041)
目的:研究戊乙奎醚光学异构体对毒蕈碱型(M)受体亚型的亲和性,为揭示戊乙奎醚的作用靶点及药效选择性提供参考。方法:利用同源建模和分子对接等分子模拟技术,通过比较戊乙奎醚不同光学异构体R1(3R,2′R)、R2(3R,2′S)、S1(3S,2′R)和S2(3S,2′S)与M受体亚型M1~M5的结合能,判断其与M受体亚型的亲和性。结果:戊乙奎醚4个光学异构体均能够对接到M受体各亚型的活性位点,不同异构体与不同M受体亚型的分子对接显示出较大差异。4个光学异构体与M3受体具有较大的结合能,在5 736.519~5 907.143 kcal/mol之间;R1与M1的结合能为1 190.04 kcal/mol;而其他光学异构体与各受体亚型的结合能较低或为负数。结论:戊乙奎醚的异构体R1对M1受体具有亲和性,4个光学异构体均对M3受体具有亲和性。
戊乙奎醚;光学异构体;毒蕈碱型受体;亚型;分子对接;同源建模
ABSTRACTOBJECTIVE:To study the affinity of penehyclidine optical isomers to muscarinic(M)receptor subtypes,and provide reference for revealing the action targets and efficacy selectivity of penehyclidine.METHODS:Homology modeling,molecular docking and other molecular simulation technologies were used to analyze and predict the binding energy of 4 optical isomers to M receptor subtypes and judge its affinity by comparing the binding energy of different optical isomers R1(3R,2′R),R2(3R,2′S),S1(3S,2′R),S2(3S,2′S)with M receptor subtypes M1-M5.RESULTS:All the 4 optical isomers can dock into the active sites of M receptor subtypes,and different optical isomers showed great differences in the molecular docking with different M receptor subtypes.Penehyclidine isomers showed larger binding energy to M3,the binding energy of 4 optical isomers ranged in 5 736.519-5 907.143 kcal/mol.The binding energy of R1 to M1was 1 190.041 kcal/mol;while those of other optical isomers to each receptor subtype were lower or negative.CONCLUSIONS:R1 shows the affinity to M1receptor.And all the 4 optical isomer show the affinity to M3.
KEYWORDSPenehyclidine;Optical isomer;Muscarinic receptor;Subtype;Molecular docking;Homology modeling
毒蕈碱型(M)受体是一个受体家族,可分为5种药理学亚型[1],即M1~M5。M1受体在中枢和外周神经系统中均有分布,参与脑部高级认知活动,如记忆、学习等,但其并非形成记忆的必需因素[2-3];同时M1受体还参与调控胃酸分泌以及与迷走神经相关的支气管收缩功能。M2受体可以调节中枢神经系统乙酰胆碱的释放[4]和心肌收缩力。M3受体则主要参与调控平滑肌收缩及腺体分泌,并可能参与食欲调控[2]。M4受体的主要作用是抑制纹状体内多巴胺的分泌,同时调节乙酰胆碱相关的运动功能以及抑制交感和副交感神经的信号传递。M5受体的作用可能与多巴胺系统有关;在虹膜、食管、淋巴细胞中也发现M5受体少量分布,但具体作用尚不清楚[5]。由于M受体各亚型的同源性和广泛分布,目前大多数抗胆碱药对不同M受体亚型选择性较差,导致了中枢和外周的各种副作用,限制了此类药物的临床应用[3]。
戊乙奎醚(Penehyclidine)是我国自行设计合成的M受体抑制剂,常用于有机磷农药中毒的急救[6],其存在4个光学异构体,即R1(3R,2′R)、R2(3R,2′S)、S1(3S,2′R)、S2(3S,2′S),以下简称R1、R2、S1、S2。药理学研究表明,不同光学异构体与M受体亚型的作用存在差异。戊乙奎醚主要选择性作用于M1和M3受体,4个光学异构体中以R1活性最强。戊乙奎醚的化学结构式见图1。
目前针对戊乙奎醚光学异构体对M受体亚型作用的研究较少。笔者在本研究中利用分子对接等计算机模拟技术研究戊乙奎醚的4个光学异构体与M1~M5受体的结合状况,为揭示戊乙奎醚的作用靶点及药效选择性提供参考。

图1 戊乙奎醚的化学结构式Fig 1 Chemical structures of penehyclidine
1 材料
高性能计算工作站(28核心56线程处理器,64 GB内存);Accelrys Discovery Studio(v 3.1)软件包;Chem Office 2014等分子模拟软件。
2 方法
2.1 配体的准备
利用Chem Office 2014软件包中的Chem Bio Draw 14.0.0.117软件绘制戊乙奎醚的分子结构,另存为标准延时格式文件(.sdf);然后利用Discovery Studio(v 3.1)软件包,将戊乙奎醚分子结构进行加氢处理,并生成三维构象和立体异构体,并进行能量最小化处理,保留能量最小化的分子构象。采用能量参数的CHARMm进行能量最小化。
2.2 M1~M4受体蛋白的准备
M1、M2、M3已有三维结构的报道,可以从蛋白质结构数据库(PDB,网址为http://www.rcsb.org)中下载。M4虽然没有完整序列的三维结构,但在PDB数据库中收载有涵盖M4序列479个氨基酸中的392个氨基酸序列的M4同源蛋白的三维结构,该结构序列涵盖了M4受体活性位点,因此也可替代M4受体进行研究。
从PDB中下载M1~M4受体蛋白的晶体数据,PDB编号分别为5CXV、3UON、4DAJ、5DSG。将上述下载的晶体文件利用Discovery Studio软件包的Prepare Protein模块进行蛋白晶体的数据的预处理,删除晶体中的水分子和非结合离子,保留蛋白链和小分子配体,修复缺失的氨基酸残基,加氢和分配电荷,并对局部环区(Loop)进行CHARMm能量最小化处理,以满足分子对接的要求。
2.3 同源建模构建M5受体蛋白
M5受体目前暂未有三维结构的报道,但具有众多的同源蛋白片段的三维结构报道,因此,可采用同源建模的方法获得M5受体的三维结构。
由于M5受体蛋白仅有一级结构的氨基酸序列已知,尚无三维结构的报道,故本文采用蛋白质建模门户网站(PMP,网址为 http://www.proteinmodelportal.org)提供的在线建模服务进行M5受体蛋白的建模,将多个建模结果进行对比,并优选出最佳的三维模型用于分子对接。
2.4 分子对接
分别利用Discovery Studio软件打开“2.2”项下预处理后的M1~M4蛋白PDB文件,选择PDB文件中的原抑制剂小分子配体,以该配体坐标为中心,半径9 Å(1 Å=10-10m)的球体范围定义为对接位点,同时将对接位点球体内的原配体分子删除。对于M5受体,本研究采用3D Ligand Site的web服务(网址为http://www.sbg.bio.ic.ac.uk/3dligandsite)来确定蛋白三维模型的配体结合位点[7],然后利用Discovery Studio将其定义为对接位点,半径同样设定为9 Å。
分子对接采用Discovery Studio软件包中的CDOCKER模块进行。载入上述经预处理并定义了对接位点的M1~M5受体蛋白分子,同时指定经过预处理的包含戊乙奎醚4个光学异构体的配体文件,指定对接位点球体坐标。对接参数设置如下:Top Hits 10;Pose Cluster Radius 0.5;Random Conformations 100;Dynamics Steps 1 000;Dynamics Target Temperature 1 000;Orientations to Refine 10;Maximum Bad Orientations 800;Orientation vdW Energy Threshold 300;Heating Steps 2 000;Heating Target Temperature 700;Cooling Steps 5 000;Cooling Target Temperature 300;Forcefield CHA-RMm;Grid Extension 8.0;Ligand Partial Charge Method Momany-Rone,Random Number Seed 314 159;Final Minimization Full Potential;Final Minimization Gradient Tolerance 0;设置并行计算,56线程。
2.5 对接的准确性判断
利用已知的蛋白-配体复合物结构,将配体小分子利用程序对接到受体蛋白,然后比较配体的实验构象与对接构象之间的差别[8-9],以均方根差(RMSD)表示。如果两种构象之间的RMSD≤2 Å,通常认为所使用的对接方法是准确可靠的,能够准确地将该受体与其配体分子进行准确对接[10]。
2.6 结合能计算
结合能(Binding energy)是表示配体与受体结合时释放出的能量,或者说将配体与受体从结合状态分离成游离状态需要提供的能量,是用来表示配体对受体亲和力的参数,一般以kcal/mol(1 kcal=4.186 8 kJ)为单位。本研究利用Discovery Studio的Calculate Binding Energies模块计算戊乙奎醚与各受体分子对接的复合物的结合能。主要参数为:Maximum Alignment RMSD 2 Å;Maximum Conformations 2 000;Electrostatics Spherical Cutoff;Nonbond List Radius 14.0;Nonbond Higher Cutoff Distance 12.0;Nonbond Lower Cutoff Distance 10.0;Partial Charge Estimation Momany-Rone。结合能越高表示受体与配体的亲和性越好,如果药物对某个亚型受体的亲和性明显高于对其他亚型受体,则说明该药物对该受体具有良好选择性。
3 结果与分析
3.1 M5受体的同源建模结果
经过PMP网站的蛋白建模服务,在所得的M5受体的三维模型中,经过对比分析,选择最佳的模型为Phyre2的模型。其三维结构见图2。
图2 同源建模所得M5受体三维结构Fig 2 3D structure of M5receptor by homology modeling
该模型采用了PDB编号为 2rh1、4rnb、3pds、3eml、3rze、4zwj、4djh、3odu、3pbl、3v2y、4grv、3vw7、3oe6、3sn6、4u16、4iar、4eiy、4ib4等 18个蛋白为同源模板,87%的残基置信区间超过95%。
69个氨基酸残基通过从头计算方式建模,配体结合位点通过Sitemap程序搜索,最终确定结合位点为ASP110、Ser114、TYR111、ASN459等残基所在的活性口袋结构。
3.2 分子对接结果
所有分子对接结果均在可接受的范围内(RMSD<2 Å),戊乙奎醚的4个光学异构体均能对接到M1受体的配体结合部位,但各异构体与M1受体蛋白活性口袋中氨基酸残基的作用模式是有差异的。配体在受体活性口袋中采取的构象不同,构象的能量就有差异,与周围氨基酸残基的作用方式不同也导致配体与蛋白的非键作用有差异,从而形成配体与受体亲和力的差异。以M1受体的分子对接为典型,结果见图3。

图3 戊乙奎醚光学异构体与M1受体的分子对接示意图Fig 3 Molecular docking diagram of penehyclidine optical isomers with M1receptor
3.2.1 戊乙奎醚光学异构体与M1受体的对接结果 光学异构体R1分子中的N原子上的孤对电子与M1受体的TYR106、TRP378、TYR404 等残基构成 P-π共轭,与ASP105残基形成盐桥作用;3位苯环与TRP378残基形成了π-π共轭的疏水作用;3位羟基则与ASN382残基形成氢键作用。R2分子中的N原子与M1受体的TYR106、SER109、TRP378、TYR404 残 基 构 成 P-π 共 轭 ,与ASP105形成盐桥作用;3位苯环与TYR381残基形成π-π共轭的疏水作用;3位羟基与ASN382残基形成氢键作用。S1分子中的N原子与TYR381、TYR404残基构成P-π共轭,与ASP105残基形成盐桥作用;3位苯环与TRP378残基构成π-π共轭;3位羟基与ASN382残基形成氢键作用。S2采取的是另一种构象,其分子中的N原子与TYR381、TYR404残基构成P-π共轭,与ASP105残基形成盐桥作用;3位羟基与ASN382残基形成氢键;3位苯环与TRP157残基构成π-π共轭。
3.2.2 戊乙奎醚光学异构体与M2受体的对接结果 4个光学异构体对接到M2受体活性口袋中,基本上为以下作用模式:均以N原子与ASP103残基形成盐桥作用,与TRP403、ASN404残基形成氢键作用;3位羟基与ASH404残基形成氢键作用。异构体间作用模式差异表现在N原子与不同氨基酸残基的共轭作用不同,主要表现为R1与TRP400、TYR104残基构成P-π共轭;R2除了TRP400和TYR104残基之外,可与TYR403残基构成P-π共轭;而S1的N原子则仅与TYR403残基构成P-π共轭,同时3位苯环未与残基有π-π共轭;S2的N原子共轭作用情况与S1类似,但3位苯环与TYR104残基构成π-π共轭。
3.2.3 戊乙奎醚光学异构体与M3受体的对接结果 光学异构体R1与R2主要通过N原子与ASP147残基形成盐桥作用,与TYR529和TYR148残基构成P-π共轭;3位羟基与ASN507残基形成氢键,与M3活性口袋中的残基发生作用。除此之外,R1的3位苯环与TYR503残基构成了π-π共轭,而R2则无此作用。S1、S2与R1、R2的结合模式不同在于N原子与TYR529和TYR506残基有P-π共轭,且3位苯环未显示π-π共轭。
3.2.4 戊乙奎醚光学异构体与M4受体的对接结果 4个异构体在M4活性空腔内均以3位羟基与ASN417残基形成氢键作用;不同的是,3位苯环和N原子与空腔内残基的作用有所区别。其中R1的3位苯环与TRP413残基构成π-π共轭,N原子与TRP413、TYR113及TYR439残基构成P-π共轭。R2除了和R1存在同样的作用外,N原子还与SER116残基形成氢键作用。S1的不同之处在于N原子与TYR439和TYR416残基构成P-π共轭,同时还与ASP112残基形成盐桥作用。S2则是N原子与TYR439、TRP413、TYR416残基构成P-π共轭,3位苯环未见与周围残基的共轭作用。
3.2.5 戊乙奎醚光学异构体与M5受体的对接结果 光学异构体R1在M5受体蛋白活性空腔内3位羟基与ASN459残基形成氢键作用;N原子与SER114残基形成氢键作用,与TYR111残基构成P-π共轭,与ASP110残基形成盐桥作用。R2作用模式为N原子与TYR111和TRP455残基构成P-π共轭,与ASP110残基形成氢键作用。S1的3位羟基与ASN459残基形成氢键作用,N原子与TYR458、TYR111残基构成P-π共轭,与ASP110残基形成盐桥作用;3位苯环取代基与TRP162残基形成π-π共轭的疏水作用。S2则是通过N原子与TYR458、TRP455TYR111残基构成P-π共轭,同时与ASP110残基形成氢键作用;3位苯环取代基与TYR459残基形成π-π共轭的疏水作用。
3.2.6 结合能计算结果 戊乙奎醚各光学异构体与M受体各亚型蛋白的分子对接的结合能计算结果见表1。
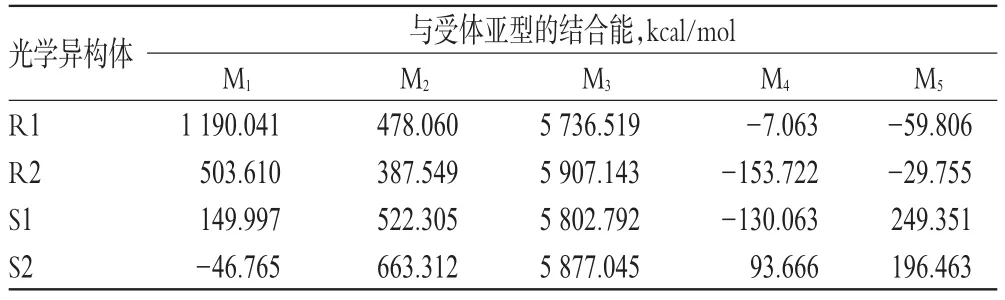
表1 戊乙奎醚光学异构体与M受体亚型的结合能计算结果Tab 1 Calculation results of binding energy of penehyclidine optical isomers to M receptor subtypes
由表1可以看出,戊乙奎醚与M3受体具有较大的结合能,4个光学异构体与M3的结合能在5 736.519~5 907.143 kcal/mol之间,远高于其他M受体亚型,预示各光学异构体均对M3受体具有较强的亲和性,各异构体均可能是有效的M3受体抑制剂。而对M1受体,各异构体表现了较大的差异,其中R1与M1的结合能最大,为1 190.04 kcal/mol;而S2与M1的结合能最小,为-46.765 kcal/mol,意味着S2与M1的结合是耗能过程,需要外界提供能量;R2、S1与M1的结合能较小,且有一定的差异,分别为503.610、149.997 kcal/mol。以上结果说明R1可能是有效的M1受体抑制剂,R2和S1可能仅有较弱抑制作用,而S2可能对M1受体无抑制作用。对于M4受体,R1、R2和S1结合能均为负值,仅S2为正值,但较小,仅为93.666 kcal/mol,说明戊乙奎醚对M4受体可能没有抑制作用或仅有极微弱的抑制作用。对M5受体,R1和R2结合能为负值,S1和S2结合能仅为249.351、196.463 kcal/mol,说明R1和R2可能对M5受体没有抑制作用,S1和S2可能仅有较微弱的抑制作用。对M2受体,各异构体均表现出较弱的亲和力,其结合能在387.549~663.312 kcal/mol之间。
从戊乙奎醚光学异构体对各M受体亚型的结合能对比来看,R1与M3结合能最大,M1其次,M2较弱,M4和M5结合能为负值,即R1对M受体的各亚型表现出明显选择性,对M3受体亲和性最强,其次是M1受体,对M2受体亲和力较弱,而对M4和M5受体无亲和性。R2和S1则仅对M3受体显示较强亲和性,对M1和M2受体仅有微弱亲和性,对M4和M5受体无亲和性。S2则对M3受体亲和性较强,对M4和M5受体亲和性较微弱,而对M1受体无亲和性。
4 讨论
小分子药物发挥作用的结构生物学基础是药物分子结合到特定受体蛋白的活性位点,促使受体蛋白的功能激活或丧失,从而产生特定的生物学效应。戊乙奎醚有2个手性碳原子,即2个手性中心,因此共有4种光学异构体。这些异构体分子量一样,因此均能进入M受体各亚型的活性空腔中,但在活性空腔中需要采取不同的构象才能与周围残基作用。一方面,对于同一个受体亚型,不同异构体由于存在手性中心的原因,其分子基团的朝向不同,需要采取不同构象与残基结合,因此分子体系的能量就有差异。通过分子对接与结合能的计算来描述这些差异,可揭示光学异构体活性差异发生的原因。另一方面,通过比较同一异构体分子对M受体不同亚型的结合能,就可以揭示或预测该化合物对M受体亚型的选择性。分子对接与结合能计算结果表明,戊乙奎醚光学异构体R1对M1和M3受体具有较好的亲和性,这与文献[11]报道的“戊乙奎醚主要对M1和M3受体有作用,对M2受体作用较弱”的结论基本一致。戊乙奎醚对M1受体的结合能计算结果显示R1与其亲和性最强,S2最弱,这一结果也与文献[12]报道一致。结合能计算结果显示戊乙奎醚对M4和M5受体亚型无亲和性或仅具有微弱亲和性,尽管未见相关药理学报道。
综上,本文利用同源建模和分子对接的手段对戊乙奎醚光学异构体与M受体各亚型进行了分子模拟研究,部分结果与已知实验结果一致。本方法可为后续研究提供一定的参考依据。
[1] Caulfield MP,Birdsall NJ.International Union of Pharmacology.ⅩⅦ.Classification of muscarinic acetylcholine receptors[J].Pharmacol Rev,1998,50(2):279-290.
[2] Eglen RM.Muscarinic receptor subtype pharmacology and physiology[J].Prog Med Chem,2005(43):105-136.
[3] Wess J.Novel insights into muscarinic acetylcholine receptor function using gene targeting technology[J].Trends Pharmacol Sci,2003,24(8):414-420.
[4] Langmead CJ,Watson J,Reavill C.Muscarinic acetylcholine receptors as CNS drug targets[J].Pharmacol Ther,2008,117(2):232-243.
[5] Mutschler E,Moser U,Wess J,et al.Muscarinic receptor subtypes:pharmacological,molecular biological and therapeutical aspects[J].Pharm Acta Helv,1995,69(4):243-258.
[6] 胡东芳,徐内卫,黄萍,等.盐酸戊乙奎醚与氯解磷定治疗急性有机磷农药中毒疗效观察[J].中国药房,2006,17(16):1246-1247.
[7] Wass MN,Kelley LA,Sternberg MJ.3DLigandSite:predicting ligand-binding sites using similar structures[J].Nucleic Acids Res,2010,38(13):469-473.
[8] Durrant J,Amaro R,Mccammon J.AutoGrow:a novel algorithm for protein inhibitor design[J].Chem Biol Drug Des,2009,73(2):168-178.
[9] Morris GM,Goodsell DS,Halliday RS,et al.Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function[J].J Comput Chem,1998,19(14):1639-1662.
[10] Akrimah,Tjahyono DH,Musadad A.Docking of potent anticancer agents;4-(pyrazol-4yl)-pyrimidine derivatives as selective cyclin-dependent kinase 4/6 inhibitors[J].International Journal of Chemical Engineering&Applications,2013,4(6):419-422.
[11] 蔡大升,裴凌.盐酸戊乙奎醚的作用机制及临床应用[J].军医进修学院学报,2008,29(2):152-154.
[12] 魏君,娄艳红,王旭光,等.高效液相色谱手性流动相添加剂法测定富马酸(R,R)-戊乙奎醚中3个光学异构体杂质的含量[J].中国药学杂志,2013,48(2):136-139.
Study on the Molecular Docking of Penehyclidine Optical Isomers and Muscarinic Receptor Subtypes
ZHANG Lijuan1,BAI Lan1,SHI Jianyou1,HE Jun2
(1.Dept.of Pharmacy,Sichuan Academy of Medical Sciences/Sichuan Provincial People’s Hospital,Chengdu 610072,China;2.Laboratory of Biotherapy,Cancer Center,West China Hospital,Sichuan University,Chengdu,610041,China)
R914.2
A
1001-0408(2017)25-3506-05
2016-12-13
2017-06-09)
(编辑:刘明伟)
DOI10.6039/j.issn.1001-0408.2017.25.14
四川省医学科学院/四川省人民医院青年科研基金(No.30305030606);四川大学-泸州市人民政府战略合作资金项目(No.2015CDLZ-S10);电子科技大学中央高校基本科研业务(No.ZYGX2015J14)。
*主管药师,硕士。研究方向:药物分析。E-mail:3998911@qq.com
#通信作者:助理研究员,博士。研究方向:小分子药物。E-mail:Jun_He@scu.edu.cn
