桑黄素和乙酰化白藜芦醇对沙奎那韦在大鼠体内药代动力学的影响
2017-09-23张敬茹于小桐李佳朋赵立波
张敬茹,于小桐,孙 宁,李佳朋,秦 一,顾 健,赵立波
(1. 北京大学人民医院药剂科,北京 100044;2. 首都医科大学附属北京儿童医院药剂科,北京 100045;3. 北京大学医学部药学院药事管理与临床药学系,北京 100191)
桑黄素和乙酰化白藜芦醇对沙奎那韦在大鼠体内药代动力学的影响
张敬茹1,3,于小桐1,3,孙 宁2,李佳朋3,秦 一2,顾 健1,赵立波2
(1. 北京大学人民医院药剂科,北京 100044;2. 首都医科大学附属北京儿童医院药剂科,北京 100045;3. 北京大学医学部药学院药事管理与临床药学系,北京 100191)
目的评估桑黄素和乙酰化白藜芦醇对大鼠体内沙奎那韦(P-gp作用底物)药物代谢动力学的影响。方法将SD大鼠分为4组,每组5只,即对照组、实验Ⅰ和Ⅱ组、阳性对照组,分别灌胃给予30 mg·kg-1沙奎那韦;30 mg·kg-1沙奎那韦联用40 mg·kg-1桑黄素;30 mg·kg-1沙奎那韦联用40 mg·kg-1乙酰化白藜芦醇和30 mg·kg-1沙奎那韦联用40 mg·kg-1维拉帕米。采用HPLC-MS/MS方法测定给药后不同时间沙奎那韦的血药浓度,计算药动学参数。结果4组大鼠体内主要药动学参数分别为:AUC0-t:381.53 μg·h·L-1,185.53 μg·h·L-1,360.43 μg·h·L-1,529.95 μg·h·L-1;AUC0-∞:409.48 μg·h·L-1,228.52 μg·h·L-1,446.67 μg·h·L-1,552.41 μg·h·L-1;Cmax:110.80 μg·L-1,86.44 μg·L-1,139.84 μg·L-1,423.60 μg·L-1;Tmax:0.25 h,0.25 h,0.25 h,0.50 h;T1/2:5.72 h,5.94 h,6.78 h,3.78 h;MRT0-∞:10.30 h,9.61 h,12.30 h,4.89 h;CL/F:7.59 mL·kg-1·h-1,13.88 mL·kg-1·h-1,7.28 mL·kg-1·h-1,5.52 mL·kg-1·h-1。结论沙奎那韦的药时曲线存在多峰现象;桑黄素可明显降低沙奎那韦的口服生物利用度并对其药动学参数产生影响,而乙酰化白藜芦醇对沙奎那韦的口服生物利用度和药动学参数没有明显影响。
桑黄素;乙酰化白藜芦醇;沙奎那韦;药动学;HPLC-MS/MS;大鼠;P-gp
目前,由人免疫缺陷病毒(human immunodeficiency virus, HIV)引起的获得性免疫缺陷综合症(acquired immune deficiency syndrome, AIDS)已经成为威胁人类生命健康的最大传染病之一。世界卫生组织推荐的高活性抗逆转录病毒疗法可明显改善艾滋病毒感染者的发病率和死亡率。其中,沙奎那韦(saquinavir, SQV)是一种HIV-1蛋白酶抑制剂,能明显抑制病毒载量,增加HIV-1患者的CD4+细胞数,目前已成为HIV-1感染治疗一线药物。与其他蛋白酶抑制剂一样,沙奎那韦可以通过竞争性地结合于病毒蛋白酶的活性位点,抑制HIV蛋白酶的活性,在被感染的细胞中产生不成熟的无感染性的病毒颗粒,使病毒不能正常装配,从而达到抑制病毒复制的目的。然而,SQV的生物利用度较低,个体间差异较大,使其治疗效果有限。在健康志愿者中,单独给予沙奎那韦的口服生物利用度只有大约4%,而变异系数却大于100%[1]。其较低的生物利用度可归因于多种因素,包括溶解度差、CYP3A介导的首过效应和肠道P-gp介导的转运外排[2]。最近的一项研究表明,肠道P-gp外排转运蛋白是导致沙奎那韦口服生物利用度低和高变异性的主要因素,而肠道CYP3A只扮演一个次要角色[3]。一方面,P-gp既能与药物结合,又能与ATP结合,ATP供能使细胞内药物泵出细胞外,降低了细胞内的药物浓度使细胞产生耐药性,影响药物在体内的组织分布和代谢过程[1]。另一方面,药物对P-gp的活性和蛋白质表达进行调控,使药物在体内产生间接的相互作用。
有研究报道[4],黄酮类、芪类化合物通过调节多药耐药基因的转录表达,影响P-gp发挥外排作用。黄酮类化合物桑黄素(morin, MOR)普遍存在于桑科植物和中草药中,具有抗肿瘤、抗炎及抗氧化作用,是近些年研究的热点。芪类化合物乙酰化白藜芦醇(acetyl-resveratrol, ACR)广泛存在于花生、葡萄等日常生活常见的植物和水果,以及葡萄酒中[5]。因此,艾滋病病患者如果同时服用含有这两种化合物的食品或药品可能对沙奎那韦的药效产生影响。Choi等[6]、Li等[7]分别报道,桑黄素可以明显增加紫杉醇和依托泊苷的药时曲线下面积和达峰浓度;Choi等[8]研究证明,白藜芦醇可以通过抑制P-gp,提高尼卡地平的药时曲线下面积和达峰浓度;实验室前期体外研究也已证明,桑黄素和乙酰化白藜芦醇可以抑制P-gp外排沙奎那韦,从而使细胞内SQV的积聚量增多[9]。已有多篇文献报道证实维拉帕米(verapamil, VER)对P-gp的抑制作用[10]。因此,基于课题组前期成果,本研究以维拉帕米为阳性对照,进一步探索桑黄素和乙酰化白藜芦醇对沙奎那韦体内药动学的影响,并为进一步评价其对药物外排转运体P-gp的影响提供实验支持,为促进药物临床合理应用、提高治疗药物浓度、增强艾滋病的治疗效果提供有价值的依据。
1 材料
1.1药品与试剂沙奎那韦标准品(SP ROCKVILLE, MD, 纯度>99%);利托那韦标准品(纯度>99%,厦门市亨瑞生化有限公司);桑黄素标准品(纯度>99%,南通飞宇生物技术有限公司);乙酰化白藜芦醇标准品(纯度>99%,杭州广林生物医药有限公司);维拉帕米注射液(规格:每支5 mg/2 mL,批号20140110,上海禾丰制药有限公司)。SQV、MOR、ACR、VER的化学结构式见Fig 1。试剂甲醇、乙腈、甲基叔丁基醚、四硼酸钠、甲酸铵和甲酸均为色谱分析纯,购自Fisher科学有限公司(美国);超纯水(Millipore公司Direct-Q®超纯水系统)。1.2仪器AB SCIEX QTRAP5500型三重四级杆串联质谱仪,配备电喷雾离子源(Turbo Ionspray)及Analyst Software(1.6)数据采集软件(美国Applied Biosystems公司);L20-A液相色谱系统(包括LC20-AT型二元输液泵、SIL-20A型自动进样器、CTO-20A型柱温箱、DGU-20A3型真空脱气机、CBM-20A型系统控制器),日本岛津公司;G20型医用离心机(北京白洋医疗器械有限公司);SORVALL Legend MICRO 21R型微型高速离心机(美国Thermo Fisher Scientific公司);VORTEX GENIE 2 型涡旋仪(美国Scientific Industries);MTN-2800D型氮吹浓缩装置(天津奥特赛恩仪器有限公司)。
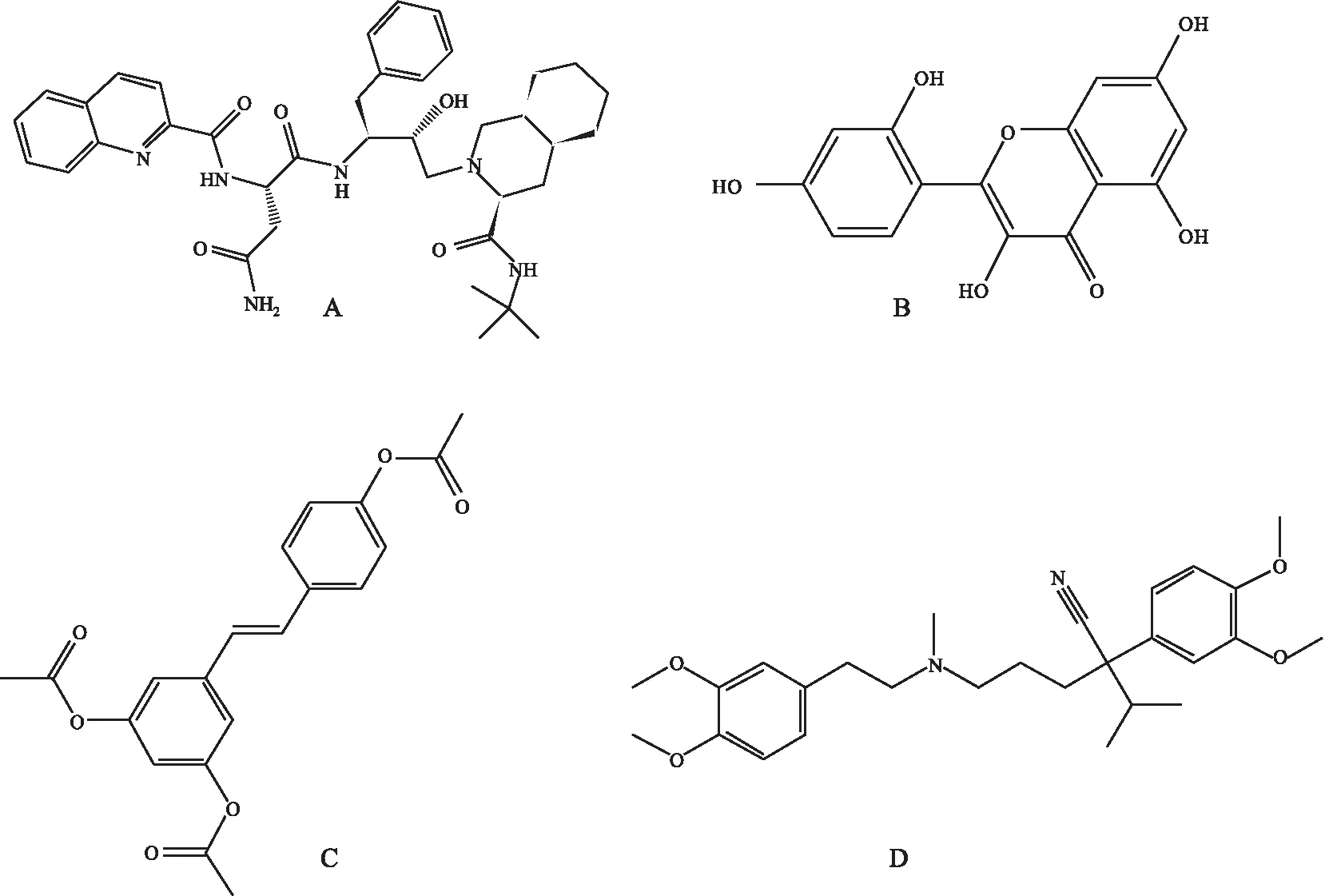
Fig 1 Chemical structures of saquinavir (A), morin (B), acetyl-resveratrol (C) and verapamil (D)
1.3实验动物健康Sprague-Dawley(SD)大鼠,6~10周,250~270 g,SPF级,♂。购自北京维通利华实验动物技术有限公司。实验前,大鼠在SPF动物实验室饲养1周。于实验前12 h禁食,实验过程中自由饮水。该研究按照“实验动物的护理和使用指南”设计,并经北京大学人民医院伦理委员会的审查和批准。
2 方法
2.1色谱条件分析柱:C18反向色谱柱(5.0 μm,150 mm×2.1 mm,美国Restek公司);预柱:HYPERSIL GOLD(3.0 μm,10 mm×2.1 mm,Thermo Scientific);流动相A:CH3CN/H2O,1 ∶99(V/V),含有0.2 mmol·L-1NH4COOH;流动相B:CH3CN/H2O,90 ∶10(V/V),含有0.2 mmol·L-1NH4COOH;流速:流动相以25 ∶75(A ∶B,V/V)比例混合,采用300 μL·min-1等度洗脱;进样量:10 μL;柱温:20 ℃。
2.2质谱条件离子源:电喷雾正离子(ESI+)检测模式;扫描方式:多重反应监测(MRM);离子喷射电压:4 500 V;温度:500 ℃;驻留时间:200 ms;源内气体1(GS1, N2)压力:40 psi;源内气体2(GS2, N2)压力:40 psi;气帘气体(N2)压力:30 psi;沙奎那韦和内标利托那韦的解簇电压(DP)分别为:0 V和0 V;碰撞能量(CE)分别为:26 V和46 V;用于定量分析监测离子对:m/z 671.4→570.4(沙奎那韦,[M+H]+),m/z 721.4→296.2(利托那韦,内标,[M+H]+)。
2.3溶液的配制
2.3.1沙奎那韦标准溶液的配制 精密称定沙奎那韦对照品10.00 mg,加甲醇-水(50 ∶50,V/V)配制成1、2、5、10、20、50、80、100 μg·L-1的标准溶液,并配制3 μg·L-1(低)、10 μg·L-1(中)、80 μg·L-1(高)的沙奎那韦质控标准溶液。
2.3.2内标(利托那韦)标准溶液的配制 精密称取利托那韦对照品10.00 mg,加甲醇-水(50 ∶50,V/V)配制成浓度为10 μg·L-1的内标溶液待用。各标准系列溶液及内标溶液均以相应体积、经过校正的容量瓶配制,并在4 ℃冰箱内保存备用。
2.3血浆样品的处理取血浆样品10 μL,依次加入200 μL甲醇-水(50 ∶50,V/V),10 μg·L-1的内标溶液200 μL,0.1 mmol·L-1四硼酸钠溶液500 μL,涡旋混合3 min后,加入甲基叔丁基醚3 mL,经涡旋混合10 min后萃取,离心5 min(4 000 r·min-1),取上层有机相2.8 mL于40 ℃下氮气吹干,残渣以200 μL流动相25 ∶75(A ∶B,V/V)复溶,取10 μL进样分析。
2.4药代动力学研究20只健康SD大鼠,随机平行分为4组:对照组、阳性对照组、实验Ⅰ和Ⅱ组。实验前12 h禁食,自由饮水,大鼠在给药4 h后自由饮食。沙奎那韦(溶剂:20%乙醇,30%丙二醇,50%生理盐水)按30 mg·kg-1剂量灌胃给药,桑黄素、乙酰化白藜芦醇和维拉帕米(混悬于生理盐水)均按40 mg·kg-1剂量灌胃给药,大鼠给药4 h后,灌胃给予2 mL生理盐水。分别于给药后5 min、15 min、30 min、1 h、1.5 h、2 h、3 h、4 h、6 h、8 h、12 h、24 h眶静脉取血200 μL,收集到肝素化的1.5 mL EP管中。血样在5 000 r·min-1条件下离心10 min,分离得到血浆约100 μL,存于EP管中,置-20 ℃冰箱储存并用于HPLC-MS/MS分析。
2.5统计学分析将血浆样品按“2.3”项下方法处理后,进行测定分析,记录色图谱,采用内标法以峰面积计算各时间点血浆药物浓度。采用WinNonlin 6.4软件的非房室模型(NCA模型)对大鼠体内的药时曲线进行拟合,计算药代动力学参数。采用IBM SPSS Statistics 20.0软件对各组药动学参数进行统计学分析,AUC0-t、AUC0-∞符合正态分布,作单因素方差分析;将CL/F、T1/2、MRT0-∞、Cmax、Cmax1、Cmax2、Cmax3进行对数转换符合正态分布,作单因素方差分析;其余参数对数转换后不符合正态分布,作非参数秩和检验。
3 结果
3.1方法学验证
3.1.1选择性 从沙奎那韦与内标利托那韦的典型多重反应监测(MRM)色谱图可知:该方法对于沙奎那韦及内标利托那韦选择性高,空白鼠血浆中的内源性物质不干扰沙奎那韦的测定,可满足定量测定要求,见Fig 2。

3.1.3精密度与准确度 SQV的日内精密度均小于12.16%,日间精密度均小于7.18%,准确度在0.89%~6.24%之间(Tab 1),说明本方法准确、可靠、重现性好。

Tab 1 Intra-day and inter-day precision and accuracy ofLC-MS/MS determination of saquinavir in rat plasma(±s, n=5)
3.1.4稳定性 经实验研究,SQV血浆样品室温放置24 h稳定(RSD在4.23%~7.10%之间),血浆样品反复3次-20 ℃、室温冻融稳定(RSD在4.93%~7.20%之间),-20 ℃冰冻放置30 d稳定(RSD在2.73%~6.32%之间)。RE均小于20%,RSD均小于15%,说明分析过程中SQV血浆样品稳定性良好。
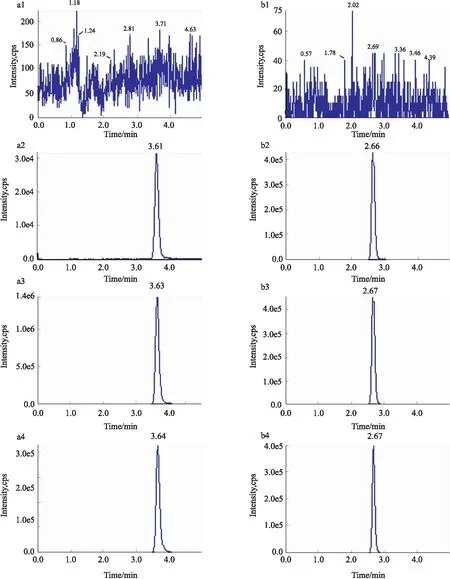
Fig 2 Representative MRM chromatograms of saquinavir(a) and ritonavir (b) in rat plasma
1: Blank plasma sample; 2: Blank plasma sample spiked with saquinavir at LLOQ (1.0 μg·L-1) and IS; 3: Blank plasma sample spiked with saquinavir at MQC (10.0 μg·L-1) and IS; 4: A plasma sample from rats collected at 4 h after oral administration of 30 mg·kg-1of saquinavir.
3.2大鼠血浆中SQV的药代动力学
3.2.1SQV血药浓度-时间曲线 SD大鼠口服单独给予沙奎那韦(30 mg·kg-1)及同时给予桑黄素、乙酰化白藜芦醇或维拉帕米(均为40 mg·kg-1)后,各组的血浆药物浓度-时间曲线见Fig 3。由图可知,各组大鼠体内沙奎那韦的药时曲线存在着较大的个体间变异。4组大鼠SQV的药时曲线均存在多峰现象。SQV对照组、SQV-MOR组、SQV-ACR组的血药浓度首次达峰时间均为服药后15 min,而SQV阳性对照组(SQV-VER组)的血药浓度首次达峰时间为服药后30 min;第2次达峰时间均为服药后1.5 h;第3次达峰时间分别为服药后8、8、8、6 h。SQV与MOR、ACR或VER同服时,前者的药时曲线发生了改变。与对照组相比,同服VER后,SQV 3次达峰浓度均升高,其中Cmax1、Cmax差异有统计学意义(P<0.05);同服MOR后,SQV 3次达峰浓度均降低,其中Cmax3差异有统计学意义(P=0.034);同服ACR后,SQV第1次达峰浓度升高,后两次达峰浓度均降低,差异均无统计学意义。

Fig 3 Mean plasma concentration-time curves of SQV after oraladministration of SQV (30 mg·kg-1) with or without morin (40 mg·kg-1) or acetyl-resveratrol (40 mg·kg-1)or verapamil (40 mg·kg-1) in rats(±s, n=5)
3.2.2药代动力学参数 大鼠口服给药后不同时间SQV的血药浓度测定结果采用WinNonlin的NCA模型进行计算,得到SQV的药代动力学参数见Tab 2。结果表明,与对照组相比,阳性对照组的AUC0-t、AUC0-∞、Cmax、Cmax1、Cmax2明显升高(P<0.05),CL/F、MRT0-∞明显降低(P<0.05),口服生物利用度升高了38.90%;同服MOR后,AUC0-t、AUC0-∞、Cmax3明显降低(P<0.05),而CL/F明显升高(P<0.05),口服生物利用度降低了51.37%;同服ACR后,对SQV的药动学参数未产生明显影响。结果说明,同时服用VER可以提高SQV的口服生物利用度,同服MOR可以降低SQV的口服生物利用度,并影响其药动学性质;但ACR与SQV同服对后者的生物利用度及药动学过程影响不明显。
4 讨论
本研究主要发现同时服用黄酮类化合物MOR可以导致SQV的口服生物利用度明显降低。在本研究中,可以观察到各组大鼠体内SQV的药时曲线存在着较大的个体间变异,这与之前的研究结果相一致[11]。尽管存在个体间差异,MOR实验组中SQV生物利用度的明显降低,表明同时服用MOR可以影响沙奎那韦在大鼠体内的药动学过程。
Tab 2 Pharmacokinetic parameters of SQV after oral administration of SQV (30 mg·kg-1) with orwithout MOR (40 mg·kg-1) or ACR (40 mg·kg-1) or VER (40 mg·kg-1) in rats(n=5,±s)
Tmax,Tmax1,Tmax2,Tmax3are presented as median (interquartile range). Relative bioavailability (RB/%) = AUCcoadmin/AUCcontrol×100.*P<0.05vsSQV group.
SQV是P-gp转运体的作用底物,具有高亲和性。小肠上皮细胞中的P-gp将SQV从小肠细胞中主动转运回肠腔,导致SQV浓度系统性的降低。有文献报道[13-14],根据P-gp作用底物的不同,黄酮类化合物鸡豆黄素A对P-gp产生抑制或者促进的不同作用。类似的现象也出现在其他黄酮类化合物的报道中,如槲皮素、山奈酚等。已有报道[11],在鼠体内MOR可以抑制P-gp外排作用,明显增加非布索坦的药时曲线下面积和达峰浓度,结合本研究结果,认为MOR可根据作用底物的不同对P-gp产生不同的调节作用。在本研究中,MOR可以促进肠道P-gp转运体的表达,减少SQV的吸收,达峰浓度降低,进而导致SQV口服利用度的明显降低。而MOR在体内外作用结果不一,推测可能是在大鼠体内MOR会刺激TNF-α/PKCβ2/NF-κB信号通路(已有文献证明该信号通路的存在),从而增加P-gp转运蛋白的表达,但仍需进一步实验证实。
此外,同时服用MOR组SQV的系统清除率(CL/F)明显升高。可观察到SQV一旦进入体循环就会迅速消除,这与较高的肝提取和胆汁排泄结果相一致[11]。胆汁排泄是SQV从机体排泄的主要途径,而肝脏P-gp在SQV排入胆汁的过程中扮演着重要角色,因此,可以推断MOR不仅可以促进介导SQV外排的小肠P-gp的表达,而且可以刺激肝脏P-gp的表达,从而导致SQV系统清除率的升高。
同时,本研究表明ACR对SQV的口服生物利用度和在大鼠体内的药动学过程影响不明显。已有研究证明,除肠道P-gp主要引起SQV的药动学性质发生变化外,细胞色素P450酶的代谢作用也可导致其发生变化。有报道发现,一方面,芪类化合物白藜芦醇在体外实验中对P-gp存在抑制作用;另一方面,白藜芦醇可能对细胞色素P450酶产生诱导作用[15]。因此,服用ACR可能抑制肠道P-gp的外排作用,增加小肠吸收,使SQV在短时间内达峰浓度升高;同时ACR可能诱导细胞色素P450酶活性增加,增加代谢作用,后两次达峰浓度降低,导致最终SQV的生物利用度没有发生明显改变,进而导致与体外实验结果不同。但SQV与ACR是否存在相互作用及其机制还需进一步实验证实。
本研究的第2个重要发现是SQV血药浓度的多峰现象。之前已有多篇文章报道了SQV在人体和大鼠体内药动学研究中类似的多峰现象,其中,肝肠循环、肠道不同部分的差异吸收和变化的胃排空是公认可以解释多峰现象的潜在机制[16]。因静脉注射SQV后不存在多峰现象,并且本研究中第2吸收相发生在大鼠恢复进食前,所以推测不是肝肠循环或胃排空引起多峰现象。一方面,溶解度是限制SQV肠道吸收的关键因素。SQV呈弱碱性,溶解度随pH增加而降低,进入小肠时会发生沉淀,进入大肠(盲肠)pH为5时,溶解度增加,吸收也随之增加;另一方面,在人类和啮齿动物中,从小肠近端到远端P-gp水平逐渐增加,在回肠表达最高。如在空肠和结肠中P-gp作用底物(依托泊苷和地高辛)的排出率比在回肠中明显降低。因此,肠道不同部分的差异吸收可能是SQV出现多峰现象的重要原因。但具体机制尚需进一步研究。
本研究表明MOR可以减少SQV的口服生物利用度,并对后者在大鼠体内的药动学过程产生明显影响;而ACR对SQV的药动学过程影响不明显,对临床HIV-1感染者治疗时同服含MOR、ACR的药物食物具有一定参考作用。
(致谢:本实验完成于首都医科大学附属北京儿童医院临床研究中心药代实验室,感谢给予帮助的老师和同学!)
[1] Wacher V J, Silverman J A, Zhang Y, et al. Role of P-glycoprotein and cytochrome P450 3A in limiting oral absorption of peptides and peptidomimetics[J].JPharmSci, 1998,87(11): 1322-30.
[2] Mouly S J, Paine M F, Watkins P B. Contributions of CYP3A4, P-glycoprotein, and serum protein binding to the intestinal first-pass extraction of saquinavir [J].JPharmacolExpTher, 2004,308(3):941-8.
[3] Usansky H H, Hu P, Sinko P J. Differential roles of P-glycoprotein, multidrug resistance-associated protein 2, and CYP3A on saquinavir oral absorption in Sprague-Dawley rats [J].DrugMetabDispos, 2008,36(5):863-9.
[4] 王玉璘,王少峡,郭 虹,等.血脑屏障中 P-糖蛋白的调节机制[J]. 中国药理学通报, 2011,27(9):1196-200.
[4] Wang Y L, Wang S X, Guo H, et al. Regulatory mechanisms of P-glycoprotein at the blood-brain barrier [J].ChinPharmacolBull, 2011,27(9):1196-200.
[5] 陈前昭,曾于桦,邵 英,等.白藜芦醇抑制人结肠癌细胞增殖与p38 MAPK的关系研究[J]. 中国药理学通报, 2016,32(8): 1100-4.
[5] Chen Q Z, Zeng Y Y, Shao Y, et al. Anti-proliferation effect of resveratrol and p38 MAPK in human colon cancer cells [J].ChinPharmacolBull, 2016,32(8):1100-4.
[6] Choi B C, Choi J S, Han H K. Altered pharmacokinetics of paclitaxel by the concomitant use of morin in rats [J].IntJPharm, 2006,323(1-2):81-5.
[7] Li X, Yun J K, Choi J S. Effects of morin on the pharmacokinetics of etoposide in rats [J].BiopharmDrugDispos, 2007,28(3):151-6.
[8] Choi J S, Choi B C, Kang K W. Effect of resveratrol on the pharmacokinetics of oral and intravenous nicardipine in rats: possible role of P-glycoprotein inhibition by resveratrol [J].Pharmazie, 2009,64(1):49-52.
[9] 李佳朋,韦忠娜,刘 阳,等. 体外研究黄酮类和芪类化合物对血脑屏障P-糖蛋白外排沙奎那韦的影响[J]. 中国临床药理学杂志, 2016,32(23):2187-90.
[9] Li J P, Wei Z N, Liu Y, et al. Effects of several flavonoids and stilbenes on P-glycoprotein mediated efflux of saquinavir on blood-brain barrierinvitro[J].ChinJClinPharmacol, 2016,32(23):2187-90.
[10] Nakagami T, Yasui-Furukori N, Saito M,et al. Effect of verapamil on pharmacokinetics and pharmacodynamics of risperidone:invivoevidence of involvement of P-glycoprotein in risperidone disposition [J].ClinPharmacolTher, 2005,78(1):43-51.
[11] Buchanan C M, Buchanan N L, Edgar K J,et al. Pharmacokinetics of saquinavir after intravenous and oral dosing of saquinavir: hydroxybutenyl-beta-cyclodextrin formulations [J].Biomacromolecules, 2008,9(1):305-13.
[12] Sahu K, Siddiqui A A, Shaharyar M, et al. Pharmacokinetic interaction between febuxostat and morin in rats[J].ExpertOpinDrugMetabToxicol, 2014,10(3):307-12.
[13] Zhang S, Sagawa K, Arnold R D,et al. Interactions between the flavonoid biochanin A and P-glycoprotein substrates in rats:invitroandinvivo[J].JPharmSci, 2010,99(1):430-41.
[14] An G, Morris M E. Effects of the isoflavonoid biochanin A on the transport of mitoxantroneinvitroandinvivo[J].BiopharmDrugDispos, 2010,31(5-6):340-50.
[15] Chow H H, Garland L L, Hsu C H,et al. Resveratrol modulates drug-and carcinogen-metabolizing enzymes in a healthy volunteer study [J].CancerPrevRes(Phila), 2010,3(9):1168-75.
[16] Davies N M, Takemoto J K, Brocks D R,et al. Multiple peaking phenomena in pharmacokinetic disposition [J].ClinPharmacokinet, 2010,49(6):351-77.
Effectsofco-administrationwithmorinandacetyl-resveratrolonpharmacokineticsofsaquinavirinrats
ZHANG Jing-ru1,3, YU Xiao-tong1,3, SUN Ning2, LI Jia-peng3,QIN Yi2, GU Jian1, ZHAO Li-bo2
(1.DeptofPharmacy,PekingUniversityPeople’sHospital,Beijing100044,China; 2.DeptofPharmacy,BeijingChildren’sHospitalAffiliatedtoCapitalUniversityofMedicalSciences,Beijing100045,China; 3.DeptofPharmacyAdministrationandClinicalPharmacy,PekingUniversityHealthScienceCenter,Beijing100191,China)
AimTo assess the impact of morin and acetyl-resveratrol on the oral bioavailability and pharmacokinetics of saquinavir (SQV), a substrate of P-glycoprotein (P-gp), in rats.MethodsTwenty rats were randomized into four groups of equal size, including a control group, two intervention groups and a positive control group, and administered orally 30 mg·kg-1SQV with or without 40 mg·kg-1morin or acetyl-resveratrol or verapamil (as positive control). The plasma concentrations of saquinavir were determined using a high-performance liquid chromatography-tandem mass spectrometry (HPLC-MS/MS) method, and the PK of SQV was assessed using non-compartmental analysis.ResultsThe PK parameters values of SQV, SQV+morin, SQV+acetyl-resveratrol, SQV+verapamil were as follows: AUC0-t, 381.53 μg·h·L-1,185.53 μg·h·L-1, 360.43 μg·h·L-1, 529.95 μg·h·L-1; AUC0-∞, 409.48 μg·h·L-1, 228.52 μg·h·L-1,446.67 μg·h·L-1, 552.41 μg·h·L-1;Cmax, 110.80 μg·L-1, 86.44 μg·L-1, 139.84 μg·L-1, 423.60 μg·L-1;Tmax, 0.25 h, 0.25 h, 0.25 h, 0.50 h;T1/2, 5.72 h, 5.94 h, 6.78 h, 3.78 h; MRT0-∞, 10.30 h, 9.61 h, 12.30 h, 4.89 h; CL/F, 7.59 mL·kg-1·h-1, 13.88 mL·kg-1·h-1, 7.28 mL·kg-1·h-1, 5.52 mL·kg-1·h-1.ConclusionsMultiple peak phenomenon can be observed in the plasma SQV profiles. Morin can significantly reduce the SQV oral bioavailability and affect SQV PK profiles while acetyl-resveratrol cannot significantly affect the SQV oral bioavailability and SQV PK profiles in rats.
morin; acetyl-resveratrol; saquinavir; pharmacokinetics; HPLC-MS/MS; rats; P-gp
10.3969/j.issn.1001-1978.2017.10.017
A
:1001-1978(2017)10-1414-07
R-332;R284.1;R969.1;R978.7
时间:2017-9-5 9:26 网络出版地址:http://kns.cnki.net/kcms/detail/34.1086.R.20170905.0925.034.html
2017-05-27,
2017-07-24
国家自然科学基金资助项目(No 81102877)
张敬茹(1993-),女,硕士生,研究方向:药代动力学,E-mail:jrzhang27@163.com; 顾 健(1962-),女,硕士,主任药师,研究方向:临床药学及药代动力学,通讯作者,E-mail:gujian201302@sina.cn; 赵立波(1979-),男,博士,主任药师,研究方向:临床药学及药代动力学,通讯作者,E-mail:libozhao2011@163.com
