线性B细胞表位预测方法研究进展①
2017-09-21羊红光
程 华 成 彬 羊红光
(河北省科学院生物研究所,石家庄050081)
线性B细胞表位预测方法研究进展①
程 华 成 彬②③羊红光③
(河北省科学院生物研究所,石家庄050081)
在抗原抗体的结合反应中,抗体参与结合的部位称抗体的对位(Paratope),抗原参与结合的部位称抗原的表位(Epitope)[1]。表位就是抗原中能被免疫细胞特异性识别的线性片段或空间构象性结构,是引起免疫应答和免疫反应的基本单位。
表位是蛋白质抗原性的基础,在蛋白的特异性抗体制备、疫苗的计算机辅助设计,特别是近期具有很大潜力的肿瘤免疫疗法中都具有非常重要的作用[2]。掌握了表位的规律将有助于绘制抗原表位图谱、降低蛋白质药物的免疫原性以及设计无毒副作用的人工疫苗等。近几年以来,随着免疫学理论、生物物理学及计算机技术的进步和广泛应用,蛋白质B细胞抗原表位的研究方法有了新的进展,一些适应于蛋白质抗原表位研究的实验检测和理论预测方法相继被发现[3,4]。本文就线性B细胞表位预测方法及相关数据库的最新进展作一综述,并指出当前存在的问题和未来发展的方向。
1 抗原表位的分类
目前,抗原表位有两种分类方法[5]:一种是按与抗原受体细胞结合不同,分为B细胞抗原表位和T细胞抗原表位;另一种是按表位结构不同分为连续性抗原表位和不连续性抗原表位。前者又称线性表位,是由肽链上顺序连续的氨基酸组成;后者又称构象抗原表位,是由那些空间邻近但顺序上不连续的氨基酸组成。线性表位可见于T细胞表位和部分B细胞表位,构象抗原表位只见于B细胞抗原表位。
2 线性B细胞表位预测方法
早期的线性B细胞表位预测基本上都是根据20种氨基酸的理化性质、结构特点及统计学等指标来进行的。人们通过观察抗原表位与已知氨基酸序列的蛋白质的某些关系,发现一些蛋白质的序列或结构特征与形成表位有关,于是提出了单一参数预测及多参数复合预测等方法。
2.1单一经典参数预测
2.1.1可及性(Accessibility) 指蛋白质中氨基酸残基被溶剂分子接触的可能性,反映了蛋白质内、外各层残基的分布情况。1979年Janin[6]统计了20种氨基酸在22个不同蛋白质中的位置数据,发现极性氨基酸更容易暴露在蛋白质的外表,而非极性氨基酸则多位于蛋白质的内部。在结合自由能的基础上,根据氨基酸在蛋白质内部和外表的位置频率,作者得到了判断某个氨基酸构成表位的几率数据。
2.1.2亲水性(Hydrophilicity) 一般认为蛋白质抗原各氨基酸残基可分为亲水性残基和疏水性残基两类,疏水性残基多埋在蛋白内部,而亲水性残基则位于蛋白表面,因此蛋白的亲水部位与蛋白抗原表位有密切的联系。常用的有Hopp-Woods参数和HPLC亲水参数。1981年Hopp等[7]做了开创性的工作,他们以6个多肽残基为一个研究单元,以有机相环境转移到水相环境的自由能为依据,计算出各种氨基酸的亲水性数据。利用这些数据能够对蛋白做出亲水性曲线,连续的高峰区则是潜在的表位区域。5年后Parker等[8]利用高效液相色谱技术重新对20种氨基酸的亲水性进行了测定,他们检测了每种氨基酸的保留时间,以此为依据计算出了不同氨基酸的亲水性参数。
2.1.3抗原性(Antigenicity) 1985年Welling等[9]通过对20个已知表位的蛋白质中氨基酸进行统计分析,得到每种氨基酸出现在抗原区的频率,将此频率除以各氨基酸在所有蛋白质中的频率就可得到一个比值,即抗原性参数。该研究发现疏水性氨基酸残基对抗原表位形成亦有贡献。1990年Kolaskar等[10]统计了已知34个蛋白质中的156个长度不超过20个氨基酸的线性表位。以氨基酸的统计数据为基础,结合亲水性、可及性及柔韧性,作者计算出了每种氨基酸位于蛋白质表面的概率。每种氨基酸位于表位的概率除以位于蛋白质表面的概率,即得到抗原特性值Ap。
2.1.4可塑性(Flexibility) 蛋白质多肽链骨架有一定程度的活动性,抗原抗体相结合时常涉及构象上的相互契合,活动性强的氨基酸残基即可塑性大的位点,易形成抗原表位。Karplas等[11]根据31个已知结构蛋白质在不同温度下多肽链骨架的变形程度,将氨基酸分为可塑性和不可塑性两大类,在参考前后氨基酸碳链可塑性对中间氨基酸的影响后,发展出了一种预测蛋白质片段活动性的方法。
2.1.5二级结构预测 (Secondary structure) 蛋白质的二级结构α螺旋、β片层及β转角等空间构象也可用于表位预测。一般认为β转角多位于蛋白质表面,利于与抗体结合,较可能成为表位。而α螺旋和β片层结构位于内部,较难嵌和抗体,不易形成表位。1993年Pellequer等[12]根据氨基酸在4种不同空间结构中的频率,得出β转角参数。
2.2多个经典参数复合预测 通过比较上述各种单参数的预测,发现正确率均不是太高。随后研究人员逐渐提出了各自不同的综合分析方法,以期提高预测的准确率。1988年Jameson和Worf[13]提出一种综合预测方案。该方案选取了4种参数,并且对各参数赋予了不同的权重,即二级结构预测是40%,亲水性参数是30%,可及性和柔韧性参数各占15%。作者利用该方案,对多个蛋白质进行了表位预测,得到了较为理想的结果。1997年万涛等[14]综合了5种参数,即Hopp亲水性、HPLC亲水性、可及性、抗原性方案和二级结构参数。作者利用含有169个已知抗原位点的34个蛋白质对这5种参数进行综合测试,得出的结论是Hopp亲水性参数和可及性参数在预测中占有重要位置,权重分别能够达到0.36,而HPLC亲水性也是比较重要的一个参数,权重达到0.18。利用这个综合预测模型,作者对5个蛋白进行了预测,准确率接近50%。
2000年Alix[15]提出了抗原性指数Antigenic Index(AG),并且使用C+语言编写了一个基于DOS操作系统的预测软件PEOPLE(Predictive estimationof protein linear epitopes)。他采用了与Jameson[13]相同的4个参数及权重。不同的是每种参数中,作者都选取了多个文献中的数据,所以他的结果要比Jameson的更加全面。2004年Saha等[16]使用7种单参数(亲水性参数、可及性、柔韧性、表面特征、极性特征、β转角参数和抗原性参数)建立了多种不同的组合,结果发现以柔韧性为主辅以亲水性及极性特征的组合,预测的准确率最高,达到58.7%。同时作者编写了一个预测软件BcePred供其他研究人员免费使用。Blythe等[17]利用50个蛋白质表位检验了AAindex数据库中484个氨基酸参数,发现即使结合了最好的参数,比随机法也仅有少许提高。多参数综合虽然比单个参数的预测准确率提高了一些,但结果依然不能让大家感到满意。
2.3基于机器学习算法的预测 随着21世纪初多种机器学习算法的出现,研究人员将这些方法引入到表位预测的工作中,即机器学习方法尝试从一套学习样本中抽提出表位的共同特点,概括为一个分类算法。结果表明准确率较传统方法有一定的提高,将B细胞表位预测方法引入了一个新的方向。
2006年4月Larsen等[18]将两种经典参数HPLC亲水性、二级结构参数与一种机器学习算法--隐马尔科夫模型(HMM,hidden Markov model)相结合,提出预测方法Bepipred。隐马尔科夫模型是一种统计分析模型,能够从可观察的参数中确定该过程的隐含参数,然后利用这些参数来作进一步的分析。Larsen方法的AROC评分达到0.671,比随机预测法和基于参数的方法有一定提高。4个月后Saha等[19]提出了基于人工神经网络模型的B细胞表位预测(Artificial neural network basedB-cell epitope prediction server,ABCpred)。人工神经网络模型是一种运算模型,是由大量处理单元互联组成的并行分布式、非线性、自适应信息处理系统。Saha的系统利用了Bcipep数据库中700个非冗余B细胞表位和Swiss-Prot数据库的700个长度为10~20个氨基酸残基的随机多肽为训练对象,在预测长度为16个氨基酸的表位时,其总体准确率接近66%。
2007年Chen等[20]首次使用机器学习算法(Amino acid pair,AAP),他们利用Bcipep数据库中872个已知B细胞表位和872个非表位随机多肽进行训练,结果表明单独使用AAP时,准确率为71%。而在结合了传统参数如亲水性、可及性等参数后,其准确率可以达到72.5%。2008年Leslie和Manzalawy等[21,22]将4种字符串函数引入到机器学习算法支持向量机(Support vector machine,SVM)中,这四种字符串函数分别是光谱函数、错配函数、局部对比函数和超序列函数。支持向量机是一种监督学习模型,可以分析数据,识别模式,用于分类和回归分析。Manzalawy利用Bcipep数据库中701个已知B细胞表位和701个非表位随机多肽进行训练,结果表明当超序列函数与SVM相结合时,准确率能达到75.8%。作者分别编写了2种不同的程序BCPred和FBCPred来进行固定长度表位(12、14、16、18和20个氨基酸)及可变长度表位的预测。
2009年Sweredoski和Baldi[23]推出了COBE pro,一个分两步来预测线性B细胞抗原表位的系统。第一步该系统对待测蛋白进行表位分析,找出所有潜在的表位,第二步则对找到的所有表位逐一进行分值运算,按分值高低进行排序。经过与现有的几种算法进行比较后作者发现该系统具有一定的竞争力。同时能够对所预测的表位按分值进行排序的特点将非常有助于使用者进行表位筛选工作。2011年Wang等[24]提出了一个新的预测体系Linear Epitope Prediction System (LEPS),该体系结合了SVM和其他相关数据,使用了4个不同的数据库(AntiJen,HIV,PC和AHP)来进行训练。在与现有4种预测方法(BepiPred,ABCPred,BCPred和FBCPred)进行比较后,作者发现LEPS预测特异性最高能达到88.33%,准确率方面最高能达到73.81%,具有明显的优势。
2013年Lin等[25]提出BEEPro,该方法将包括氨基酸比例参数、位置特异性得分矩阵(Position specific scoring matrix,PSSM)及14种氨基酸物理化学特性在内的16种参数与Support Vector Machine (SVM)相结合。作者使用了6个不同的数据库(Sollner、AntiJen-1、AntiJen-2、HIV、Pellequer和PC)来训练该体系。最终作者认为多理化参数预测的结果明显要好于单个参数的结果。同时,在与现有5种已知预测方法(LEPS、BepiPred、ABCPred、BCPred和FBCPred)进行比较后,作者发现BEEProS预测特异性是94.33%~99.46%,准确率为93.73%~99.29%,具有非常明显的优势。2个月后Singh等[26]首次使用全部经过实验验证的49 694个表位序列和50 324个非表位多肽序列进行数据的训练,并将这些数据分为了5个包括固定长度和可变长度不同的数据库。作者认为与以往使用随机多肽作为非表位肽进行训练的方法相比,他们使用经实验确定的非表位多肽序列,将得到更为可信的预测结果。
2014年Lian等[27]提出了基于多元线性回归的表位预测(Epitope prediction using the multiple linear regression,EP MLR),一种基于MLR的线性表位预测系统。作者从BEOracle中选取了300个线性表位和300个随机多肽作为训练对象,在与现有3种预测方法(ABCPred、BCPred和LEPS)进行比较后,作者发现EP MLR在预测特异性和准确率方面与这些预测方法基本一致。2015年Shen等[28]提出了一种将a mino acid anchoring pair composition(APC)和SVM两种机器学习相结合的预测系统APCpred。作者使用了6个表位数据库对该系统进行训练,分别是BC1727、Chen872、ABC16、Blind387、Lbtope-Confirm及FBC934。在与现有3种预测方法(ABCPred、BCPred和LEPS)进行比较后,作者发现APCpred在预测特异性和准确率方面与这些预测方法基本一致。
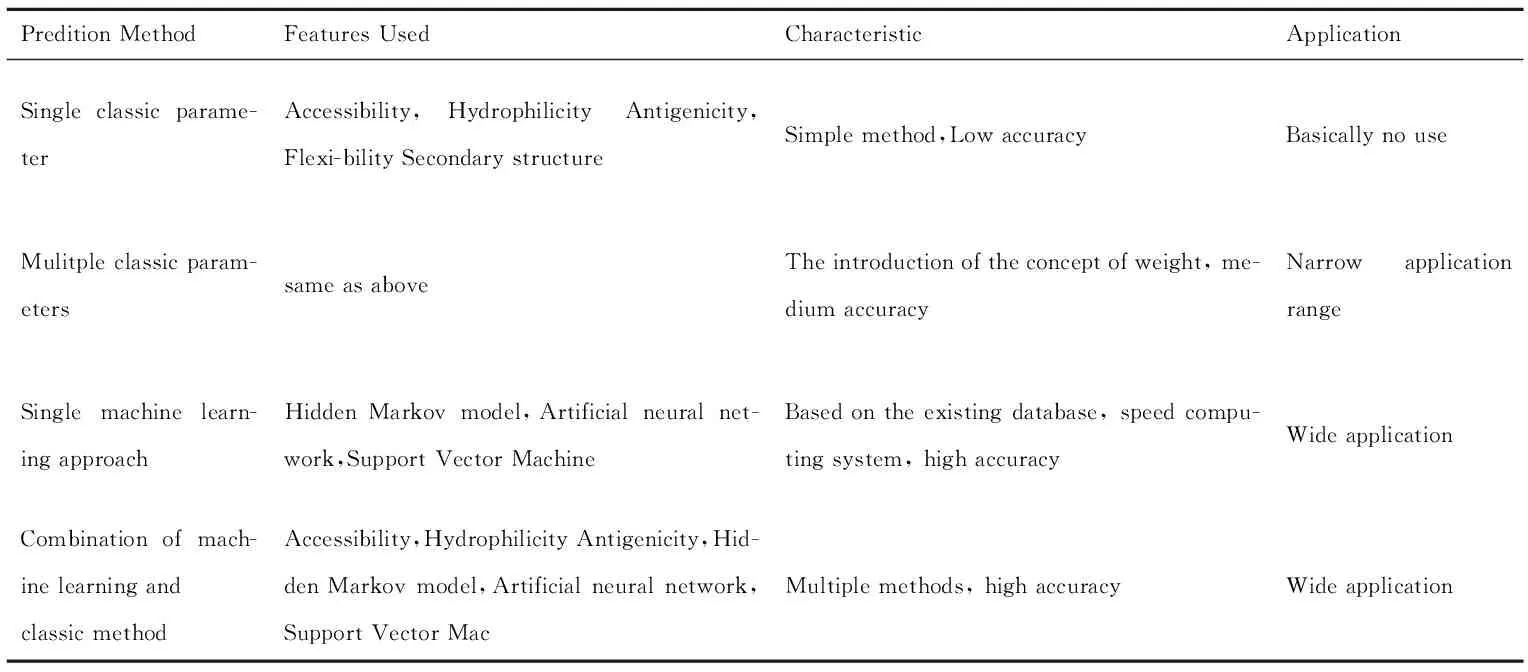
2.4各种预测方法比较 对目前各种预测方法进行了比较,见表1。
3 线性B细胞表位在线预测网站
目前多个研究小组在互联网上发布了线性表位的预测网址,总结见表2。
表1各种预测方法比较
Tab.1Compareofdifferentpredictionmethods
PreditionMethodFeaturesUsedCharacteristicApplicationSingleclassicparame-terAccessibility,HydrophilicityAntigenicity,Flexi-bilitySecondarystructureSimplemethod,LowaccuracyBasicallynouseMulitpleclassicparam-eterssameasaboveTheintroductionoftheconceptofweight,me-diumaccuracyNarrowapplicationrangeSinglemachinelearn-ingapproachHiddenMarkovmodel,Artificialneuralnet-work,SupportVectorMachineBasedontheexistingdatabase,speedcompu-tingsystem,highaccuracyWideapplicationCombinationofmach-inelearningandclassicmethodAccessibility,HydrophilicityAntigenicity,Hid-denMarkovmodel,Artificialneuralnetwork,SupportVectorMacMultiplemethods,highaccuracyWideapplication
表2线性B细胞表位预测网址
Tab.2WebsiteoflinearB-cellepitopesprediction

TimeNameWebsiteReference2004BcePredhttp://www.imtech.res.in/raghava/bcepred/[16]2006BepiPredhttp://www.cbs.dtu.dk/services/BepiPred/[18]2006ABCpredhttp://www.imtech.res.in/raghava/abcpred/[19]2007VaxiJenhttp://www.jenner.ac.uk/VaxiJen[29]2008BCPREDShttp://ailab.ist.psu.edu/bcpred/pre-dict.html[21-22]2009COBEprohttp://scratch.proteomics.ics.uci.edu[23]2009Epitopiahttp://epitopia.tau.ac.il/[30]2011LEPShttp://leps.cs.ntou.edu.tw[24]2012IEDBhttp://tools.iedb.org/main/bcell/[31]2013LBtopehttp://crdd.osdd.net/raghava/lbtope/index.php[26]2014EPMLRhttp://www.bioinfo.tsinghua.edu.cn/epitope/EPMLR/[27]2015APCpredhttp://ccb.bmi.ac.cn/APCpred/pre-diction.php[28]
4 小结与展望
表位是蛋白质抗原性的基础,也与生物机体免疫调节密切相关,表位研究对疾病的诊断以及疫苗分子的设计等具有重要的意义。经过多年的研究积累,特别是近10年来机器学习算法和高速计算机技术的帮助,表位预测研究有了突飞猛进的发展,新预测方法和数据库层出不穷。尽管如此,表位预测的总体表现尚不能令人十分满意,多个方面仍需进一步改进和提高。相信随着对蛋白质功能和结构研究的不断深入,抗原表位及蛋白质3D结构数据库的不断丰富及生物信息学技术等学科的快速发展,表位预测的准确性也将进一步提升,人们对蛋白质抗原表位的研究将更加透彻。
[1] Van Regenmortel MH.The concept and operational definition of protein epitopes[J].Philos Trans R Soc Lond B Biol Sci,1989,323(1217):451-466.
[2] Daga DA,Davila ML.CAR models:next-generation CAR modificationsfor enhanced T-cell function[J].Mol Ther Onco,2016,3:16014-16021.
[3] Correia BE,Bates JT,Loomis RJ,etal.Proof of principle for epitope-focused vaccine design[J].Nature,2014,507(7491):201-206.
[4] Uhlen M,Bandrowski A,Carr S,etal.A proposal for validation of antibodies[J].Nature Methods,2016,13(10):823-827.
[5] Barlow DJ,Edwards MS,Thornton JM.Continous and discontinuous protein antigenic determinants[J].Nature,1986,322(6081):747-748.
[6] Janin J.Surface and inside volumes in globular proteins[J].Nature,1979,277:491-492.
[7] Hopp TP,Woods KR.Prediction of protein antigenic determinants from a mino acid sequences[J].Proc Natl Acad Sci,1981,78(6):3824-3828.
[8] Parker JMR,Guo D,Hodges RS.New hydrophilicity scale derived from high-performance liquid chromatography peptide retention data:correlation of predicted surface residues with antigenicity and X-ray-derived accessible sites[J].Biochemistry,1986,25:5425-5432.
[9] Welling GW,Weijer WJ,Vander ZR,etal.Prediction of sequential antigenic regions in proteins[J].FEBS Lett,1985,188(2):215-218.
[10] Kolaskar AS,Tongaonkar PC.A semi-emperical method for prediction of antigenic determinants on protein antigens[J].FEBS Lett,1990,276(1):172-174.
[11] Karplus PA,Schultz GE.Prediction of chain flexibility in proteins:a tool for the selection of peptide antigen[J].Naturwissenschaften,1985,72(4):212-213.
[12] Pellequer JL,Westhof E,Van Regenmortel MH.Correlation between the location of antigenic sites and the prediction of turns in proteins[J].Immunol Lett,1993,36(1):83-99.
[13] Jameson BA,Wolf H.The antigenic index:a novel algorithm for predicting antigenic determinants[J].Bioinformatics,1988,4(1):181-186.
[14] 万 涛,孙 涛,吴加金.蛋白顺序性抗原决定簇的多参数综合预测[J].中国免疫学杂志,1997,13(6):329-333.
[15] Alix AJ.Predictive estimation of protein linear epitopes by using the program PEOPLE[J].Vaccine,1999,18(3-4):311-314.
[16] Saha S,Raghava G.BcePred:Prediction of continuous B-cell epitopes in antigenic sequences using physico-chemical properties[J].Int Conference on Artificial Immune System,2004,3239:197-204.
[17] Blythe MJ,Flower DR.Benchmarking B cell epitope prediction:underperformance of existing methods[J].Protein Sci,2005,14(1):246-248.
[18] Larsen JEP,Lund O,Nielsen M.Improved method for predicting linear B-cell epitopes[J].Immunome Res,2006,2(1):2-9.
[19] Saha S,Raghava G.Prediction of continuous B-cell epitopes in an antigen using recurrent neural network[J].Proteins,2006,65:40-48.
[20] Chen J,Liu H,Yang J,etal.Prediction of linear B-cell epitopes using a mino acid pair antigenicity scale[J].Amino Acids,2007,33:423-428.
[21] Manzalawy YEL,Dobbs D,Honavar V.Predicting linear B-cell epitopes using string kernels[J].Mol Recognit,2008,21:243-255.
[22] Leslie C,Eskin E,Cohen A,etal.Mismatch string kernels for discriminative protein classification[J].Bioinformatics,2004,20(4):467-476.
[23] Sweredoski MJ,Baldi P.COBEpro:a novel system for predicting continuous B-cell epitopes[J].Protein Eng Des Sel,2009,22(3):113-120.
[24] Wang HW,Lin YC,Pai TW,etal.Prediction of B-cell linear epitopes with a combination of support vector machine classification and a mino acid propensity identification[J].J Biomed Biotechnol,2011,447-451.
[25] Lin SYH,Cheng CW,Su ECY.Prediction of B-cell epitopes using evolutionary information and propensity scales[J].BMC Bioinformatics,2013,14( 2):10-19.
[26] Singh H,Ansari HR,Raghava GP.Improved method for linear B-cell epitope prediction using antigen′s primary sequence[J].PLoS One,2013,8(5):62216-62224.
[27] Lian Y,Ge M,Pan XM.EP MIR:sequence-based linear B-cell epitope prediction method using multiple linear regression[J].BMC Bioinformatics,2014,15:414-420.
[28] Shen KW,Cao Y,Cha L,etal.Predicting linear B-cell epitopes using a mino acid anchoring pair composition[J].BioData Mining,2015,8:14-26.
[29] Doytchinova IA,Flower DR.VaxiJen a server for prediction of protective antigens,tumour antigens and subunit vaccines[J].BMC Bioinformatics,2007,8:4-11.
[30] Rubinstein ND,Mayrose I,Martz E,etal.Epitopia:a web-server for predicting B-cell epitopes[J].BMC Bioinformatics,2009,10:287-292.
[31] Kim Y,Ponomarenko J,Zhu Z,etal.Immune epitope database analysis resource[J].Nucleic Acids Res,2012,40:525-530.
[收稿2016-12-27 修回2017-01-13]
(编辑 张晓舟)
10.3969/j.issn.1000-484X.2017.09.032
①本文受河北省科学院两院合作项目(15005023)资助。
程 华(1977年-),男,硕士,副研究员,主要从事抗体工程研究。
R392.1
A
1000-484X(2017)09-1422-05
②共同第一作者。
③河北省科学院应用数学研究所,石家庄050081。
