蒽环类药物的心脏毒性机制
2017-09-12综述吴星恒审校
秦 聪(综述),吴星恒(审校)
(南昌大学a.研究生院医学部2014级; b.第一附属医院儿科,南昌330006)
蒽环类药物的心脏毒性机制
秦 聪a(综述),吴星恒b(审校)
(南昌大学a.研究生院医学部2014级; b.第一附属医院儿科,南昌330006)
蒽环类药物(ANT)是应用最广泛的抗肿瘤药物之一,但由于其严重的心脏毒性,临床应用受到限制。早期研究认为ANT的心脏毒性作用与氧化应激及铁代谢失衡有关,新近的研究发现,ANT还可通过干扰基因的表达及加快细胞自噬导致心脏损伤,本文就氧化应激、铁代谢失衡、细胞自噬、基因表达等方面,对ANT致心脏毒性机制进行综述。
蒽环类药物; 心脏毒性; 机制
蒽环类药物(ANT)应用于临床已达半世纪之久,被越来越多的化疗方案采用以发挥其最大的抗肿瘤效应,成为现代肿瘤治疗学的重大成就之一。近年来某些化疗药物逐渐被靶向治疗所替代,而ANT在肿瘤联合化疗方案中的地位仍难以取代[1-3]。由于ANT严重的心脏毒性使其临床应用受到了极大的限制,心脏毒性大小往往呈剂量相关性,与非蒽环类化疗药物联合治疗时,心脏毒性常常加重。ANT致心脏毒性作用可随时发生,即使是缓解期的肿瘤患者也不能幸免[4-5]。关于ANT致心脏毒性机制,早期研究以氧化应激、铁代谢失衡为主,近年来发现ANT致心脏毒性是多种机制共同作用的结果,并将研究重点转移到基因表达层面。本文就几种ANT致心脏毒性机制作一综述。
1 ANT产生活性氧簇(ROS)与心肌损伤
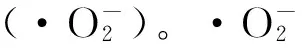
ANT产生的ROS及有毒醛类物质对多种细胞及组织均有损害作用,为什么心脏对ANT的损伤最敏感呢?首先,由于心肌细胞缺乏抗氧化酶,因此抗氧化活性较其他组织、细胞弱。肝脏中过氧化氢酶的含量是心脏的150倍,超氧化物歧化酶的含量约为心脏的4倍[7]。Doroshow等[7]研究表明尽管正常肝脏和心脏所含谷胱甘肽过氧化物酶活性相同,但阿霉素作用后心脏的谷胱甘肽过氧化物酶却更易受其影响致活性下降。实验以递增浓度的阿霉素(5、10、15 mg·kg-1)作用于小鼠,发现阿霉素剂量越高,小鼠心脏的谷胱甘肽过氧化物酶活性越低,心肌细胞损伤越严重;而心脏的超氧化物歧化酶、肝脏的超氧化物歧化酶以及肝脏的谷胱甘肽过氧化物酶活性受同一剂量的阿霉素影响不明显。由此猜想,心脏对抗过氧化物损伤主要通过谷胱甘肽过氧化物酶作用,但其活性可受阿霉素的影响而降低,使心脏缺乏对抗氢过氧化损伤及脂质过氧化损伤的能力。其次,ANT与心肌细胞有高度亲和力,Parker等[8]通过建立2H标记的心磷脂的阴离子型磷脂双分子模型,在磁场存在的条件下,阿霉素与此模型结合可导致2H的分裂,以此来证实阿霉素与心磷脂的亲和力,结果表明阿霉素对属于阴离子型磷脂双分子的心磷脂具有高度亲和力。而动物体内的心磷脂大部分存在于线粒体细胞内膜及心肌中。同时心肌细胞本身也富含线粒体,线粒体占据心肌细胞内近50%的空间。另一方面,相较于肝脏线粒体,心肌细胞的线粒体可产生额外的NADH脱氢酶,循环生成更多ANT相关自由基参与氧化应激损伤[9-10]。
2 铁代谢失调、ATP结合盒转运蛋白B8(ABCB8)与心肌损伤
Ichikawa等[11]发现在培养的新生鼠心肌细胞中,阿霉素的作用可使线粒体内铁含量显著升高,ABCB8 mRNA及其蛋白质水平却显著下降。ABCB8属于ABC蛋白家族中的一员,有研究[12-13]表明,ABCB8通过协调线粒体铁的输出对维持线粒体内铁平衡及细胞质Fe/S蛋白成熟有重要作用。由此猜测,阿霉素致心肌细胞铁失衡与其调节ABCB8有关。Ichikawa等[11]还发现在ABCB8 mRNA及蛋白质下调时,线粒体铁转运蛋白-2(一种参与线粒体内铁输入的蛋白)水平无明显改变,但与铁存储有关的线粒体铁蛋白及其mRNA水平显著上升,说明阿霉素致心肌细胞铁失衡是由于铁的输出受抑制而并非是铁输入的增加。离体实验中,阿霉素作用于ABCB8表达下调的新生鼠心肌细胞,发现线粒体中的铁水平明显增高,而细胞核或者细胞质中的铁含量无明显改变,通过MTS分析及TUNEL法测定本组的心肌细胞死亡率也较阿霉素作用的普通新生鼠心肌细胞明显升高。另一方面,使ABCB8基因过表达可抑制阿霉素所致心肌细胞线粒体铁聚集并明显减少心肌细胞死亡率,说明阿霉素致心脏损伤与其抑制ABCB8的表达进而影响心肌细胞的铁代谢有关。
3 带有caspase富集功能域的凋亡抑制因子(ARC)、MicroRNA-532-3p与心肌损伤
ARC最初是作为内源性细胞凋亡抑制因子被发现。它对内源性及外源性的细胞凋亡信号途径均有对抗作用。ARC对于保持心脏生理功能也具有重要作用,在病理条件下,ARC下调可引起多种心脏疾病的发生,如心肌肥大、心力衰竭和心肌梗死。
Wang等[14]将阿霉素作用于离体的心肌细胞,发现不论是在ARC mRNA水平还是在其蛋白水平,数量均下调。阿霉素剂量为2 μmol·L-1时,线粒体开始分裂,心肌细胞死亡率逐渐增加,在这个过程中,Caspase-3的活性明显增加。但阿霉素作用于可高表达ARC的心肌细胞时,线粒体分裂、心肌细胞凋亡及Caspase-3的活性均降低。小剂量阿霉素(0.2 μmol·L-1)作用于正常心肌细胞时,仅有一小部分心肌细胞发生线粒体分裂、细胞凋亡,而相同的小剂量阿霉素作用于敲除ARC基因的心肌细胞,线粒体分裂、细胞凋亡及Caspase-3的活性再次显著增加。由此猜测ANT致心脏毒性可能与其下调ARC表达有关。
MicroRNA(miRNA)是一类内源性的具有调控功能的非编码区的RNA,可通过控制mRNA自身的稳定及其翻译过程对靶基因进行负性调节。miRNA也具有调节心脏功能的作用,例如控制心脏电信号的传导、心肌细胞的收缩、心脏的生长及形态的改变等。Wang等[14]通过RNA杂交工程及即时定量聚合酶连锁反应(qRT-PCR)研究ARC基因的3′非编码区作用时发现,阿霉素作用使ARC表达下调时,一些miRNA数量增加,其中miRNA-532-3p数量增加明显。进一步的荧光标记法证实miRNA-532-3p可通过作用于3′非编码区来调节ARC的生成。单使心肌细胞的miRNA-532-3p扩增对线粒体的裂解及凋亡无明显影响,但加入阿霉素后,即使是小剂量(0.2 μmol·L-1),miRNA-532-3p扩增的心肌细胞也会出现明显增多的线粒体裂解及心肌细胞凋亡,说明阿霉素通过miRNA-532-3p下调ARC水平,使线粒体裂解增加进而促进心肌细胞凋亡。因为ARC在某些肿瘤细胞中也可高表达,阿霉素使肿瘤细胞中ARC表达下调的过程中是否也有miRNA-532-3p参与?在一些肿瘤细胞中(例如Hela、SGC-7901、SW-480、HepG-2),miRNA-532-3p表达水平较心肌细胞低。在Hela、SGC-7901、SW-480、HepG-2肿瘤细胞株中加入阿霉素,并未发现miRNA-532-3p的表达水平有明显改变。敲除miRNA-532-3p基因的Hela、SGC-7901、SW-480、HepG-2肿瘤细胞株在作用于阿霉素后,其细胞凋亡情况较未敲除该基因的肿瘤细胞株无明显差异。由此可见,miRNA-532-3p并未参与阿霉素的抗肿瘤过程,仅在阿霉素致心脏毒性中发挥作用。这为研究既不干扰阿霉素抗肿瘤作用,又可保护心脏的抗心肌损伤药物提供了方向。
4 心肌细胞自噬
ANT引起的心脏毒性涉及多种不同的蛋白质消耗,包括细胞自噬在内的多种蛋白降解系统参与了ANT相关心脏毒性的发生。当细胞自噬发生时,由自噬相关基因ATG8编码的微管相关蛋白质轻链3(LC3)酶解掉一小段多肽,使LC3-I(18 000)转变为LC3-Ⅱ(16 000),并进入自噬小体(AVs)形成其中一部分。因此LC3-Ⅱ的水平在一定程度上与自噬小体的数量正相关。为了解阿霉素对心肌细胞的致自噬作用,Kobayashi等[15]将心肌细胞培养在加有1%牛血清的DMEM培养基中,使用阿霉素(1 μmol·L-1)作用18 h后测定LC3-Ⅱ的水平。蛋白质印迹实验(WB)表明,阿霉素作用后LC3-Ⅱ水平显著升高。但是LC3-Ⅱ及AVs的水平会随着自噬小体的形成和降解发生变化,自噬流可以反映自噬小体转移至溶酶体的量及在其中降解的程度,进一步反映细胞自噬的活性。该实验通过比较溶酶体抑制剂存在与否的条件下AVs或LC3-Ⅱ的含量变化来反映自噬流,为了解阿霉素引起LC3-Ⅱ的聚集是因为自噬流还是因为蛋白降解系统自身功能失调,根据是否加入溶酶体抑制剂巴弗洛霉素A1(BFAA1)将阿霉素作用的心肌细胞再次分组。结果表明,加入BFAA1的阿霉素作用组较未加入BFAA1的阿霉素作用组,LC3-Ⅱ增加更显著,说明阿霉素可通过加快心肌细胞自噬引起心脏损伤。
5 心肌锚定重复序列蛋白(CARP)、转录因子GATA4与心肌损伤
CARP是经典的肌肉序列蛋白家族中的一类。在培养的大鼠心肌细胞中使用干扰性小核糖核酸(siRNA)抑制CARPmRNA转录,发现CARP减少的同时丝状肌动蛋白及肌间蛋白条纹数量均急剧下降,大量肌纤维被破坏致显著的肌节紊乱[15]。CARP对阿霉素极为敏感,Chen等[16]将阿霉素作用于体外培养的小鼠心肌细胞,发现即使0.5 μmol·L-1剂量的阿霉素足以对CARP的表达产生显著的抑制。
GATA4属于锌指结构转录因子GATA家族中的一员。GATA家族中的6个成员通过识别相应基因片段并与之结合起到调节目的基因转录的作用。GATA1、2、3在调节血细胞及血管内皮细胞基因转录中起到重要作用,GATA4、5、6在新生的心脏中表达,而GATA4在成熟的心脏中有较大的活性。GATA4作为重要的转录调节因子,对心肌细胞的分化、心肌肌节的合成及心肌细胞的存活起着重要的作用,但阿霉素可下调GATA4连接活性,引起心肌纤维破坏、肌节紊乱,进而出现心肌收缩功能减弱影响心脏功能[17]。
Park等[18]发现柔红霉素(DNR)可以抑制小鼠心肌细胞GATA4 mRNA的表达且DNR的这种作用依赖于p53基因,使用抑制剂-α(一种p53的抑制剂)可抑制DNR对GATA4的下调作用。
Chen等[16]在小鼠心肌细胞内使用siRNA敲除GATA4基因,CARP基因启动子的活性及CARP蛋白数量均下降,致大量心肌细胞肌节紊乱,使用载有GATA4基因的腺病毒转染一部分敲除GATA4基因的小鼠心肌细胞使GATA4基因过表达,24 h后将相同剂量阿霉素分别作用于GATA4过表达组及GATA4无表达组,发现较GATA4无表达组,GATA4过表达组的CARP水平明显增高,免疫组织化学显示心肌细胞M线被很好的保留,证明GATA4过表达可抑制阿霉素致心肌肌节紊乱并猜测ANT可通过下调GATA4致CARP数量减少,进而引起心肌细胞肌节紊乱。
研究发现GATA4与ANT致心肌细胞自噬也有关系。Kobayashi等[15]将阿霉素(1 μmol·L-1)作用于培养的新生鼠心肌细胞,发现LC3-Ⅱ在加入阿霉素的1~3 h就开始显著增加,随着阿霉素的加入,LC3-Ⅱ的数量一直持续上升,相反,GATA4的数量在最初作用的1~3 h减少最显著,之后持续下降。进一步研究,使心肌细胞GATA4基因过表达,在阿霉素作用下,LC3-Ⅱ数量下降,加入BFAA1的心肌细胞组也可见LC3-Ⅱ的聚集明显减少。当敲除心肌细胞GATA4基因后,可发现阿霉素致心肌细胞自噬流显著增加。另一方面,当GATA4过表达使心肌细胞自噬流减少时,Bcl-2蛋白显著增加;GATA4基因敲除后,心肌细胞自噬流显著增加,但近90%的Bcl-2蛋白随之减少。由此猜测GATA4参与的ANT致心肌细胞自噬可能是通过调节Bcl-2的表达起作用。
6 结语
ANT致心脏毒性是由多种机制共同作用引起。除了引起氧化应激及铁代谢失衡外,ANT可通过加快心肌细胞自噬作用并影响多种基因的表达,使其编码的功能蛋白发生改变从而引起心肌细胞的功能失调,最终导致心肌细胞死亡,造成心脏不可逆的损伤。
[2] Petrelli F,Borgonovo K,Cabiddu M,et al.Neoadjuvant chemotherapy and concomitant trastuzumab in breast cancer:a pooled analysis of two randomized trials[J].Anticancer Drugs,2011,22(2):128-135.
[3] Reinbolt R E,Patel R,PAN Xueliang,et al.Risk factors for anthracycline-associated cardiotoxicity[J].Support Care Cancer,2016,24(5):2173-2180.
[4] Menna P,Paz OG,Chello M,et al.Anthracycline cardiotoxici-ty[J].Expert Opin Drug Saf,2012,11( S 1):S21-36.
[5] Conway A,Mccarthy A L,Lawrence P,et al.The prevention,detection and management of cancer treatment-induced cardiotoxicity:a meta-review[J].BMC Cancer,2015,15:366.
[6] 王彦妮.醌与氧之间电子转移反应的量子化学研究[D].北京: 中国科学院化学研究所,1999.
[7] Doroshow J H,Locker G Y,Myers C E.Enzymatic defenses of the mouse heart against reactive Oxygen metabolites:alterations produced by doxorubicin[J].J Clin Invest,1980,65(1):128-135.
[8] Parker M A,King V,Howard K P.Nuclear magnetic resonance study of doxorubicin binding to cardiolipin containing magnetically oriented phospholipid bilayers[J].Biochim Biophys Acta,2001,1514(2):206-216.
[9] Nohl H,Gille L,Staniek K.The exogenous NADH dehydrogenase of heart mitochondria is the key enzyme responsible for selective cardiotoxicity of anthracyclines[J].Z Naturforsch C,1998,53(3/4):279-285.
[10] Lindsey M L,Lange R A,Parsons H,et al.The tell-tale heart:molecular and cellular responses to childhood anthracycline exposure[J].Am J Physiol Heart Circ Physiol,2014,307(10):H1379-H1389.
[11] Ichikawa Y,Ghanefar M,Bayeva M,et al.Cardiotoxicity of doxorubicin is mediated through mitochondrial Iron accumulation[J].J Clin Invest,2014,124(2):617-630.
[12] Ichikawa Y,Bayeva M,Ghanefar M,et al.Disruption of ATP-binding cassette B8 in mice leads to cardiomyopathy through a decrease in mitochondrial Iron export[J].Proc Natl Acad Sci USA,2012,109(11):4152-4157.
[13] Gammella E,Maccarinelli F,Buratti P,et al.The role of Iron in anthracycline cardiotoxicity[J].Front Pharmacol,2014,5:25.
[14] Wang J X,Zhang X J,Feng C,et al.MicroRNA-532-3p regulates mitochondrial fission through targeting apoptosis repressor with caspase recruitment domain in doxorubicin cardiotoxicity[J].Cell Death Dis,2015,6(3):e1677.
[15] Kobayashi S,Volden P,Timm D,et al.Transcription factor GATA4 inhibits doxorubicin-induced autophagy and cardiomyocyte death[J].J Biol Chem,2010,285(1):793-804.
[16] Chen B,Zhong Lin,Roush S F,et al.Disruption of a GATA4/ankrd1 signaling axis in cardiomyocytes leads to sarcomere disarray:implications for anthracycline cardiomyopathy[J].PLoS One,2012,7(4):e35743.
[17] Angsutararux P,Luanpitpong S,Issaragrisil S.Chemotherapy-Induced cardiotoxicity:overview of the roles of oxidative stress[J].Oxid Med Cell Longev,2015,2015(11):795602.
[18] Park A M,Nagase H,Liu Lingling,et al.Mechanism of anthracycline-mediated down-regulation of GATA4 in the hea-rt[J].Cardiovasc Res,2011,90(1):97-104.
(责任编辑:罗芳)
2017-02-12
R96
A
1009-8194(2017)06-0104-02
10.13764/j.cnki.lcsy.2017.06.041
