miR-101在胰腺癌组织中的表达水平及对癌细胞侵袭能力的影响*
2017-08-30李晓怡陈小丽刘水逸石玉香卢忠心陈卫群
李晓怡 陈小丽 张 琴 刘水逸 石玉香 卢忠心 陈卫群
miR-101在胰腺癌组织中的表达水平及对癌细胞侵袭能力的影响*
李晓怡1陈小丽1张 琴1刘水逸1石玉香3卢忠心1陈卫群2,#
目的:检测分析胰腺癌组织和胰腺癌细胞株中微小RNA(miRNA)的表达情况及对胰腺癌细胞侵袭能力的影响。方法:收集临床患者胰腺癌组织、癌旁组织标本及4种典藏胰腺癌细胞株,用实时荧光定量聚合酶链反应(qRT-PCR)检测所有样本中miR-101的表达水平;采用Transwell侵袭试验检测miR-101对胰腺癌细胞株PANC-1和MIA PaCa-2的侵袭能力,同时采用Western Blotting检测转染miR-101的模拟物(mimic)后胰腺癌细胞株MIA PaCa-2的Rho相关卷曲螺旋蛋白激酶2(Rhoassociated coiled coil forming protein kinas,ROCK2)蛋白表达水平。结果:4种胰腺癌细胞株中miR-101表达量较正常胰腺细胞明显降低(P<0.05),胰腺癌组织中miR-101表达量也明显低于癌旁组织(P<0.05)。转染组miR-101的胰腺癌细胞株PANC-1和MIA PaCa-2的侵袭能力较未转染组明显减弱(P<0.05),转染组胰腺癌细胞株MIA PaCa-2中ROCK2蛋白表达也显著低于未转染组(P<0.05)。结论:胰腺癌组织和胰腺癌细胞株均低表达miR-101,可能靶向抑制ROCK2蛋白,减弱胰腺癌细胞的侵袭能力。
微小RNA-101;胰腺癌;侵袭;Rho相关卷曲螺旋蛋白激酶2
胰腺癌是一种预后极差的恶性肿瘤,胰腺癌患者 5年生存率低于5%。胰腺癌的发病率在全球范围内逐年递增,已成为癌症导致死亡的4大原因之一[1, 2]。大于90%的胰腺癌患者为胰腺导管癌(Pancreatic Ductal Adenocarcinoma,PDAC),容易迅速侵犯周围组织,转移到其它的脏器,大量研究表明,上皮间质转化(Epithelial-Mesenchymal Transition,EMT)在PDAC的转移和侵袭过程中发挥着至关重要的作用[3]。由于胰腺癌发生初期的患者没有症状,呈现局部侵袭,使得胰腺癌的早期诊断困难,大约只有20%的患者有机会接受手术治疗。因此,能尽早发现和评估胰腺癌的特异性标志物,对临床诊断和选择合适的治疗方案、评估患者的预后均有重要的临床意义。已有研究表明,微小RNA(microRNA,miRNA)在肿瘤的发生、发展和凋亡过程中扮演着癌基因和抑癌基因双重作用,可能作为肿瘤诊断指标和治疗靶点。近期研究发现,miR-101在肿瘤的发生发展中扮演着至关重要的角色,miR-101通过不同途径和机制调控多种癌细胞功能,对骨肉瘤、胆囊癌、结肠癌、前列腺癌等发挥着抑癌基因的作用[4, 5],包括抑制Rho相关卷曲蛋白螺旋蛋白激酶2(ROCK2,miR-101的重要靶基因之一)表达,影响EMT进一步阻遏癌症进展。但目前有关PDAC癌组织和体外细胞株miR-101表达水平及其对癌细胞侵袭作用的实验报道较少,因此,本研究通过检测胰腺癌细胞株和胰腺癌组织中miR-101的表达情况,探讨转染miR-101对胰腺癌侵袭转移的抑制作用。
1 材料与方法
1.1 临床组织标本
收集胰腺癌组织和癌旁组织共15对[6](切取距肿瘤组织≥3cm的胰腺正常组织为癌旁组织),均为2015-10-2017-01间经病理确诊收入武汉市中心医院的患者手术切除标本,其中男8例,年龄37-67岁,平均52.0±15.0岁;女7例,年龄43-63岁,平均53.0±10.0岁。本研究经院伦理委员会批准,所有研究者均签署知情同意书。
1.2 胰腺癌细胞株
人胰腺癌细胞株PANC-1(CRL-1469)、 MIA PaCa-2(CRL-1420)、BxPC-3(CRL-1687)、AsPC-1(CRL-1682)和胰腺导管正常细胞株H6C7均购自美国典藏细胞库(ATCC)。
1.3 主要试剂和仪器
miR-101模拟物(miR-101 mimic)以及阴性对照(mimic nc)购自上海吉玛公司(B02001); miR-101逆转录试剂盒和荧光定量PCR分析试剂盒(7031、P0301、O1026)购自锐博公司;DMEM培养基(1521768)、RPMI-1640培养基(1408794)、转染用OPTI-MEM培养基(1785924)、胎牛血清(1688857)购自美国Gibco公司;RNA提取试剂TRIzol(139504)、miR-101 mimic和mimic nc转染试剂Lipofectamine LTX(1630855)购自美国Invitrogen公司;Transwell小室(14115030)购自美国Corning公司,Matrigel基质胶(4342007)购自美国BD公司;BCA蛋白测定试剂盒(MA152711)购自美国Thermo公司,ECL化学发光试剂盒(9610298)购自美国GE公司;鼠抗人β-actin单克隆抗体(E3013)、羊抗鼠IgG(E0213)和羊抗兔IgG(B1711)购自美国Santacruz公司,兔抗人ROCK2多克隆抗体(ab71598)购自美国Abcam公司。紫外分光光度(DU800)计购自美国贝克曼公司,PCR扩增仪(StepOne plus)购自美国Lifetechnologies公司。
1.4 miR-101基因水平检测
1.4.1 临床组织样本处理:从液氮中取出胰腺癌组织和癌旁组织在干冰中,迅速用匀浆器将组织碾磨成小块,临床组织标本和典藏细胞株总RNA用TRIzoL提取。
1.4.2 典藏细胞株分组培养:PANC-1、MIA PaCa-2细胞均置于DMEM培养基中,分别为P组、M组;BxPC-3、AsPC-1、H6C7细胞均置于RPMI-16BXPC-30培养基中,分别为B组、A组和H组;上述培养基中均加入1%青-链霉素和10%胎牛血清。将各组细胞移至底部面积为25cm2培养瓶中,于37℃、5% CO2培养箱中恒温培养。
1.4.3 实时荧光定量PCR(qRT-PCR):采用TRIzoL提取临床组织样本和典藏细胞株总RNA(按试剂盒说明书操作),并检测RNA纯度及浓度,取1μg总RNA与1μl逆转录酶、3μl逆转录引物混合,在15μl 锐博miRNA分析反应体系中,16℃ 30 min,42℃ 30 min,85℃ 5min,进行逆转录。然后取1.33μl逆转录产物 cDNA 与1μl引物混合,在20μl 反应体系中,95℃变性10 min;95℃ 15s、0℃ 60s,40个循环。miR-101 逆转录引物:5′-CTC AAC TGG TGT CGT GGA GTC GGC AAT TCA GTT GAG TTC AGT TAT-3′;U6逆转录引物:5′-AAC GCT TCA CGA ATT TGC GT-3′。miR-101定量上游引物:5′-ACA CTC CAG CTG GGT ACA GTA CTG TGA TAA-3′,下游引物: 5′-CTC AAC TGG TGT CGT GGA-3′;U6定量上游引物:5′-CTC GCT TCG GCA GCA CA-3′,下游引物:5′-AAC GCT TCA CGA ATT TGC GT-3′。组织样本miR-101的相对表达量用2-ΔCt来计算(以U6作内对照),典藏细胞株miR-101相对表达量用2-ΔΔCt计算(以H6C7表达量为对照)。
1.5 PANC-1、MIA PaCa-2细胞侵袭能力检测
采用Transwell小室技术[7],取对数生长期的MIA PaCa-2和PANC-1细胞接种6孔板,每株细胞接种6个复孔,待细胞密度达到60%融合后,按照Lipofectamine LTX试剂盒说明书用mimic nc和miR-101 mimic分别转染两株胰腺癌细胞(未转染组和转染组),48h后用无血清培养基重悬细胞调整至密度为5×104个细胞/ml,在提前用Matrigel基质胶包被的Transwell小室的上室中加入200μl细胞悬液,下室中加入600μl Opti-MEM培养基,培养24h后,用多聚甲醛固定小室30min,将上室内细胞擦去再用吉姆萨染液染色下室细胞,显微镜下观察计数进入下室的细胞数目,每孔计数5个视野取平均值作为该孔细胞数。发生侵袭的癌细胞呈紫蓝色。
1.6 MIA PaCa-2细胞株ROCK2蛋白检测
应用Western Blotting方法,用蛋白酶抑制剂RIPA(含1%PMSF)裂解转染后MIA PaCa-2细胞;用BCA试剂盒提取总蛋白、检测蛋白浓度,12%聚丙烯酰胺凝胶电泳2h分离ROCK2,电转移至0.22μm的PVDF膜,5%脱脂奶粉封闭1h,加入一抗lgG 4℃孵育过夜,洗涤,ROCK2二抗孵育1h;最后加入化学发光底物,成像仪扫描结果。以β-actin为参照,以目标蛋白与β-actin灰度比值作为ROCK2蛋白相对表达量。
1.7 统计学处理
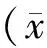
2 结果
2.1 临床胰腺癌组织和典藏细胞株miR-101基因水平
胰腺癌组织和癌旁组织中miR-101的基因水平分别为4.74±2.17、9.18±3.86,前者明显低于后者(t=3.89,P<0.05)。各典藏胰腺癌细胞株miR-101基因水平差异有统计学意义(F=81.194,P<0.01)。P组、B组、A组、M组miR-101水平均低于H组,其中M组(0.15±0.04)低于A组(0.40±0.05),A组低于B组(0.05±0.07),B组低于P组(0.76±0.10),差异均具有统计学意义,(t均≥3.93,P均<0.01),见图1。
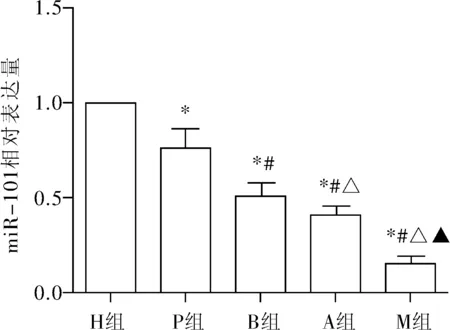
注:与H组比较,*P<0.01;与P组比较,#P<0.01;与B组比较,△P<0.01;与A组比较,▲P<0.01
2.2 转染miR-101胰腺癌细胞株侵袭能力
图2显示,无论是MIA PaCa-2细胞还是PANC-1细胞,转染组发生侵袭的细胞数目均较未转染组细胞减少。定量分析表明,转染组细胞株较未转染组细胞株侵袭细胞数目明显减少,差异有统计学意义(P<0.05),见表1。
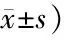
表1 转染miR-101胰腺癌细胞侵袭数目(个,
注:与未转染组同一细胞株比较,1)P<0.01
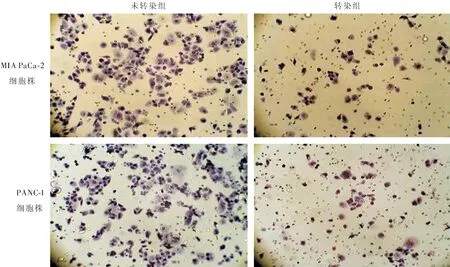
注:图中紫蓝色细胞为发生侵袭的癌细胞,转染组癌细胞株侵袭能力明显低于未转染组癌细胞株
2.3 转染miR-101胰腺癌细胞株ROCK2蛋白表达
MIA PaCa-2胰腺癌细胞株转染miR-101 mimic 24h后,Western Blotting结果显示未转染组ROCK2蛋白相对表达量为1.38±0.06,而转染组ROCK2蛋白相对表达量为0.62±0.07,明显低于未转染组(t=14.45,P<0.01),见图3。
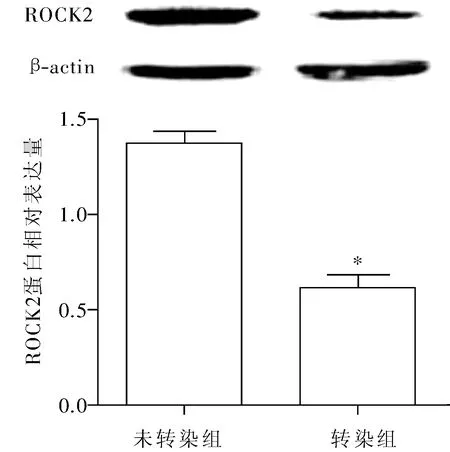
注:与未转染组比较,*P<0.01
3 讨论
自从1993年在线虫体内发现miRNA lin-4至今,已有超过数千种人类miRNA被发现并鉴定,虽然miRNA在人类基因组中所占比例只有1%-3%,但人类30%的基因却受其调控,包括在肿瘤的发生、发展和机体细胞的失控性增殖、凋亡减少、侵袭转移和血管生成等过程中扮演癌基因或抑癌基因的角色。已有大量研究证实,miR-101通过不同的生理过程监管着细胞的多个功能,可发挥着抑癌基因的作用。Wang等[8]报道骨肉瘤组织中miR-101低表达;Bao等[5]在研究胆囊癌时发现有转移的胆囊癌组织中miR-101明显低表达,且miR-101的表达水平与肿瘤的大小、TMN分级、转移程度和有无淋巴结转移密切相关。研究还发现miR-101主要通过抑制MAPK/ERK和Smad通路的激活从而抑制胆囊癌细胞的增殖侵袭能力[4, 9, 10]。本研究所选病种虽与上述不同,但结果发现与正常胰腺细胞株相比,胰腺癌细胞株的miR-101表达也明显降低,并且与临床癌旁组织相比,胰腺癌组织中miR-101也呈低表达,进一步验证了不同癌细胞的抑癌基因miR-101水平都是降低的。
本研究检测分析了癌细胞株MIA PaCa-2经转染miR-101后ROCK2蛋白水平变化。结果显示,转染miR-101的胰腺癌细胞株,其ROCK2水平明显下降,侵袭能力减弱。ROCK2属于Rho 家族的下游效应蛋白,是细胞内ROCK的两种异构体之一。可通过多种途径参与到肿瘤的远端浸润侵袭、恶性转型,包括:磷酸化各种有丝氨酸或苏氨酸残基的蛋白质底物,如细胞骨架连接蛋白(ERM)家族等调节细胞收缩或通过调节基质金属蛋白酶(MMPs)等的活性,参与降解细胞外基质,还可以促进肿瘤迁移过程中微丝介导的黏着斑形成以及肿瘤细胞的运动能力等,分别或共同促进肿瘤细胞的转移和侵袭[11-15]。所以过表达miR-101能够干预胰腺癌细胞抑制其靶基因ROCK2的蛋白表达,减弱癌细胞株的侵袭力。
综上所述,miR-101在胰腺癌细胞株和胰腺癌组织中均呈低表达,并且能靶向作用于ROCK2减弱胰腺癌细胞的侵袭能力,因此认为miR-101可能是抑制胰腺癌进展的分子之一,临床检测miR-101水平或过表达miR-101可能为胰腺癌的诊断和治疗提供新的思路,对于miR-101在抑制胰腺癌进程的具体作用机制还需进一步研究。
本文第一作者简介:
李晓怡(1991-),女,汉族,硕士,技师,主要从事miRNA参与肿瘤发生发展的机制研究
1 Ueda J, Matsuda Y, Yamahatsu K, et al. Epithelial splicing regulatory protein 1 is a favorable prognostic factor in pancreatic cancer that attenuates pancreatic metastases[J]. Oncogene, 2013,33(36):4 485-4 495.
2 Wen F, Shen A, Choi A, et al. Extracellular DNA in pancreatic cancer promotes cell invasion and metastasis[J]. Cancer Research, 2013,73(14):4 256-4 266.
3 Zhao X, Gao S, Ren H, et al. Hypoxia-Inducible factor-1 promotes pancreatic ductal adenocarcinoma invasion and metastasis by activating transcription of the actin-bundling protein fascin[J]. Cancer Research, 2014,74(9):2 455-2 464.
4 Ye Z, Yin S, Su Z, et al. Downregulation of miR-101 contributes to epithelial-mesenchymal transition in cisplatin resistance of NSCLC cells by targeting ROCK2[J]. Oncotarget, 2016, 7(25):37 524-37 535.
5 Bao R, Shu Y, Hu Y, et al. miR-101 targeting ZFX suppresses tumor proliferation and metastasis by regulating the MAPK/Erk and Smad pathways in gallbladder carcinoma[J]. Oncotarget, 2016, 7(16):22 339-22 354.
6 Aguilera KY, Rivera LB, Hur H, et al. Collagen signaling enhances tumor progression after anti-VEGF therapy in a murine model of pancreatic ductal adenocarcinoma[J]. Cancer Research, 2014,74(4):1 032-1 044.
7 Lu Z, Li Y, Takwi A, et al. miR-301a as an NF-kB activator in pancreatic cancer cells[J]. The EMBO Journal, 2011, 30(1):57-67.
8 Wang Z, He R, Xia H, et al. MicroRNA-101 has a suppressive role in osteosarcoma cells through the targeting of c-FOS[J]. Exp The Med, 2016, 11(1):1 293-1 299.
9 Liu Z, Wang J, Mao Y, et al. MicroRNA-101 suppresses migration and invasion via targeting vascular endothelial growth factor-C in hepatocellular carcinoma cells[J]. Oncology Letters, 2016, 11(1):433-438.
10 Wang H, Meng Y, Cui Q, et al. miR-101 targets the EZH2/Wnt/β-catenin the pathway to promote the osteogenic differentiation of human bone marrow-derived mesenchymal stem cells[J]. Scientific Reports, 2016,6:36 988-36 994.
11 Hsu C, Chang Z, Lee H. Immunohistochemical evaluation of ROCK activation in invasive breast cancer[J]. BMC Cancer, 2015,15(1).346-357.
12 Han Y, Wang X, Chen J, et al. Noise-induced cochlear F-actin depolymerization is mediated via ROCK2/p-ERM signaling[J]. Journal of Neurochemistry, 2015,133(5):617-628.
13 Li M, Zhou W, Yuan R, et al. ROCK2 promotes HCC proliferation by CEBPD inhibition through phospho-GSK3β/β-catenin signaling[J]. FEBS Letters, 2015,589(9):1 018-1 025.
14 Huang D, Du X, Yuan R, et al. Rock2 promotes the invasion and metastasis of hepatocellular carcinoma by modifying MMP2 ubiquitination and degradation[J]. Biochemical and Biophysical Research Communications, 2014,453(1):49-56.
15 Rath N, Morton JP, Julian L, et al. ROCK signaling promotes collagen remodeling to facilitate invasive pancreatic ductal adenocarcinoma tumor cell growth[J]. EMBO Mol Med, 2017,9(2):198-218.
Expression and Effect of miR-101 on Invasion in Patients with Pancreatic Cancer
LI Xiao-yi1, CHEN Xiao-li1, Zhang Qin1, LIU Shui-yi1, SHI Yu-xiang3, LU Zhong-xin1, CHEN Wei-qun2,#
1Department of Clinical Laboratory;2The Central Laboratory,3Department of Pathology, The Central Hospital of Wuhan, Tongji Medical College,Huazhong University of Science and Technology, Wuhan 430014, China;#
Objective: To investigate the expression of microRNA in pancreatic cancer tissue of patients and pancreatic cancer cell lines and the effect on invasion of pancreatic cancer.Method: Constitutive expression of miR-101 was detected by quantitative real-time polymerase chain reaction (qRT-PCR) in pancreatic cancer tissue and adjacent tissue, the relative expression of miR-101 was measured by qRT-PCR in 4 pancreatic cancer cells or normal pancreatic cell. The protein level of target gene ROCK2 was examined by western blot and the ability of cell invasion was detected by transwell after transfection of miR-101 in MIA PaCa-2.Results: The expression of miR-101 was downregulated in pancreatic cancer tissue and 4 pancreatic cancer cells (P<0.05). After overexpressing miR-101 in MIA PaCa-2 and PANC-1, the protein level of target gene-ROCK2 was reduced and the ability of pancreatic cell invasion was also inhibited (P<0.05).Conclusion: miR-101 was downregulated in pancreatic cancer tissue and 4 pancreatic cancer cells, miR-101 may have a great effect on pancreatic cancer cell invasion by directly target ROCK2.
miR-101; Pancreati cancer; Invasion,;Rhoassociated coiled coil forming protein kinas
湖北省自然科学基金重点项目(2015CFA078),武汉市卫计委科研项目(WX14C13,WX14B10)
华中科技大学同济医学院附属武汉中心医院,武汉 430014;1检验科;2中心实验室;3病理科;#
,E-mail: 2899919002@qq.com
本文2016-12-26收到,2017-02-23修回
R735.9
A
1005-1740(2017)03-0011-05
