药物治疗儿童肝豆状核变性肝功能失代偿的效果观察
2017-08-30李春霞龚敬宇张梅虹王建设
李春霞, 龚敬宇, 张梅虹, 王建设
(1 延安大学附属医院 感染病科, 陕西 延安 716000; 2 上海复旦大学附属金山医院 儿科, 上海 201500;3 上海复旦大学附属儿科医院 肝病中心, 上海 201102)
药物治疗儿童肝豆状核变性肝功能失代偿的效果观察
李春霞1, 龚敬宇2, 张梅虹2, 王建设3
(1 延安大学附属医院 感染病科, 陕西 延安 716000; 2 上海复旦大学附属金山医院 儿科, 上海 201500;3 上海复旦大学附属儿科医院 肝病中心, 上海 201102)
目的 观察药物治疗对以肝功能失代偿起病的肝豆状核变性患儿的影响。方法 收集2015年8月1日-2016年8月1日复旦大学附属金山医院收治的以“肝硬化,肝功能失代偿”起病的肝豆状核变性患儿3例,均经过ATP7B基因测序进一步确诊。所有患儿入院后立即给予低铜饮食和对症支持治疗,在明确肝豆状核变性的临床诊断后给予青霉胺和葡萄糖酸锌片联合治疗。记录患儿临床转归情况。结果 3例患儿均检测到ATP7B基因复合杂合突变。经内科综合治疗后,患儿低蛋白血症和凝血功能逐步改善,其中2例患儿Child-Pugh评分均有所降低。结论 对于不明原因肝功能异常甚至肝硬化患儿,诊断需常规考虑肝豆状核变性,早期诊断、及时恰当的药物治疗可减少肝移植手术需求。
肝豆状核变性; 肝硬化; 药物疗法; 儿童
肝豆状核变性又名威尔逊氏病,其世界范围内的发病率约为1/3万,致病基因携带率约1/90[1-3],是一种罕见的常染色体隐性遗传的铜代谢障碍疾病。该病由ATP7B基因突变引起铜离子蓄积于体内的肝、脑、肾、角膜等部位[4],导致各脏器进行性损伤。伊朗曾有学者[5]对106例5个月至18岁的肝硬化儿童进行平均(8.24±6.12)年的研究,发现儿童肝硬化的最常见原因即威尔逊氏病,建议对所有肝硬化儿童进行威尔逊氏病排除。但由于其临床表现多样,首发症状各异,并且随年龄改变呈现不同临床特点,故常容易出现误诊或漏诊,甚至发展为失代偿期肝硬化、肝衰竭,最终需要接受肝移植。笔者对以“肝硬化,肝功能失代偿”起病的肝豆状核变性儿童使用药物治疗,成功逆转了肝功能失代偿状态,避免了肝移植,现将结果报告如下。
1 资料与方法
1.1 研究对象 病例1:患儿男性,8岁,于2015年12月14日因“双踝关节疼痛4个月余”收入复旦大学附属金山医院。4个月前患儿无明显原因及诱因出现双踝关节疼痛不适,2个月前出现双脚踝水肿。当地医院双腿及踝关节MR平扫提示双侧小腿筋膜炎,皮下脂肪性水肿,左侧胫腓骨远端局灶性骨髓水肿,左踝关节腔内少量积液。1个月前外院查腹部MR示肝硬化并多发结节,不除外铜代谢异常。入院查体:神志清楚,对答切题,无性格行为异常,皮肤、巩膜未见明显黄染,双下肢可见散在出血点,可见肝掌,颜面部可见数枚蜘蛛痣,腹平软,肝脾肋下未及,移动性浊音阴性,双下肢不肿,生理发射存在,病理反射未引出。生长发育曲线正常。入院查角膜色素环(K-F环)阳性,头颅MR示双侧苍白球T1W1高信号,后颅窝蛛网膜囊肿可能。铜蓝蛋白<0.079 g/L,24 h尿铜0.842 mg。血常规示WBC 4.8×109/L,Hb 107 g/L,PLT 67×109/L。肝功能示TBil 18.6 μmol/L,DBil 15.1 μmol/L,TBA 178.2 μmol/L,ALT 50 U/L,AST 72 U/L,ALP 566 U/L,GGT 53 U/L,Alb 33 g/L。凝血功能示PT 30.3 s,国际标准化比值(INR)2.6,血浆纤维蛋白原(FIB)1.43 g/L,活化部分凝血活酶时间(APTT)60.4 s;经注射维生素K1后PT无缩短。
病例2:患儿男性,10岁,于2015年8月1日因“肝功能异常13 d”收入复旦大学附属金山医院。患儿13 d前无明显诱因发热,在当地医院就诊时发现肝功能异常,PLT减少,腹部B超提示肝脏弥漫性病变,门静脉增宽,脾肿大。腹部CT提示肝硬化。入院查体:神志清楚,对答切题,无性格行为异常,皮肤、巩膜无黄染,无出血点,未见肝掌及蜘蛛痣,双肺呼吸音清,无啰音,腹平软,肝肋下未及,脾肋下约8 cm,质韧,移动性浊音阴性,双下肢不肿,生理发射存在,病理反射未引出。生长发育曲线正常。入院查头颅CT未见明显病变,K-F环阳性。血铜蓝蛋白<0.078 g/L,24 h尿铜1.269 mg。血常规示WBC 1.88×109/L,Hb 102 g/L,PLT 32×109/L。肝功能示TBil 12.6 μmol/L,DBil 8.1 μmol/L,TBA 28.2 μmol/L,ALT 105 U/L,AST 114 U/L,ALP 251 U/L,GGT 193 U/L,Alb 26 g/L。凝血功能示PT 20 s,INR 1.77,FIB 0.94 g/L,APTT 34.1 s;经注射维生素K1后PT无缩短。
病例3:患儿女性,8岁,于2016年4月18日因“肝功能异常1个月余”收入复旦大学附属金山医院。1个月前患儿因间断性右侧头痛,伴有发热,在当地医院就诊,给予门诊“青霉素”治疗2 d后,头痛症状缓解,但出现双下肢水肿,腹部增大,皮肤、巩膜黄染,小便色黄,在当地医院住院13 d,查肝功能明显异常,给予保肝、利尿治疗后水肿消退,但肝功能无明显改善。为进一步诊治,来复旦大学附属金山医院就诊。入院查体:神志清楚,对答切题,无性格行为异常,皮肤、巩膜未见明显黄染,可见肝掌,未见蜘蛛痣,腹平软,肝脾肋下未及。移动性浊音阴性,双下肢不肿,生理发射存在,病理反射未引出。生长发育曲线正常。入院查K-F环阳性,头颅MR示双侧苍白球T1W1高信号,后颅窝蛛网膜囊肿可能。上腹部MR示肝硬化,腹水形成。血铜蓝蛋白<0.080 8 g/L,24 h尿铜1.394 mg。血常规示WBC 6.2×109/L,Hb 86 g/L,PLT 131×109/L。肝功能示TBil 61.2 μmol/L,DBil 45 μmol/L,ALT 126 U/L,AST 251 U/L,GGT 301 U/L,Alb 26.5 g/L。凝血功能示PT 26.1 s,INR 2.18,FIB 1.37 g/L,APTT 49.8 s。
1.2 基因诊断 经患儿及家长知情同意后,抽取外周静脉EDTA抗凝血2 ml,常规抽提DNA,用于肝豆状核变性ATP7B致病基因PCR扩增全部编码外显子并Sanger双向测序。以NM_000053.2为模板序列,查阅千人基因组(TGP)数据库、阿尔伯塔大学建立的ATP7B基因突变专用数据库(http://www.wilsondisease.med.ualberta.ca/),并结合已发表文献判断变异的致病性。新发现的变异使用MutationTaster预测致病性。
1.3 内科治疗措施 所有患儿入院后立即给予低铜饮食和对症支持治疗,在明确肝豆状核变性的临床诊断后给予青霉胺和葡萄糖酸锌片联合治疗。具体方法为:青霉素皮试阴性后,青霉胺2次/d,125 mg/次,每周随访血、尿常规,根据耐受情况逐渐加量至10 mg·kg-1·d-1,最大量不超过750 mg/d,分2次,分别在早餐前1 h和晚餐前1 h服用;葡萄糖酸锌片2次/d,5片/次(共含锌元素50 mg),分别在中餐前1 h和睡前空腹服用,根据胃肠道耐受性调整剂量,并可加量至最多每日锌元素180 mg。结合胆红素、Alb和凝血功能恢复正常后改为单用葡萄糖酸锌维持治疗,剂量按24 h尿铜定量调整。服用青霉胺期间每日补充30 mg维生素B6。并注意联合治疗时两种药物一定要分开服用,中间间隔4~6 h,防止螯合剂和锌剂相互作用,影响疗效。
1.4 观察指标 对患儿肝功能、PT及血常规等各项指标进行动态观察,以监测治疗的不良反应及治疗效果。
1.5 随访 患儿出院后,门诊根据其治疗情况进行每2周至2个月的不定期随访。
2 结果
2.1 基因诊断结果 3例患儿均检测到ATP7B基因复合杂合突变(表1),其中病例1发现的c.2333G>T、c.2310C>G、c.1531C>T,病例2发现的c.2333G>T、c.3029A>C,病例3发现的c.1639C>T均为已知致病性突变[6-11];而病例3中发现的c.2356-2delA突变未见文献报道,但预测其严重影响剪切,为致病性突变。
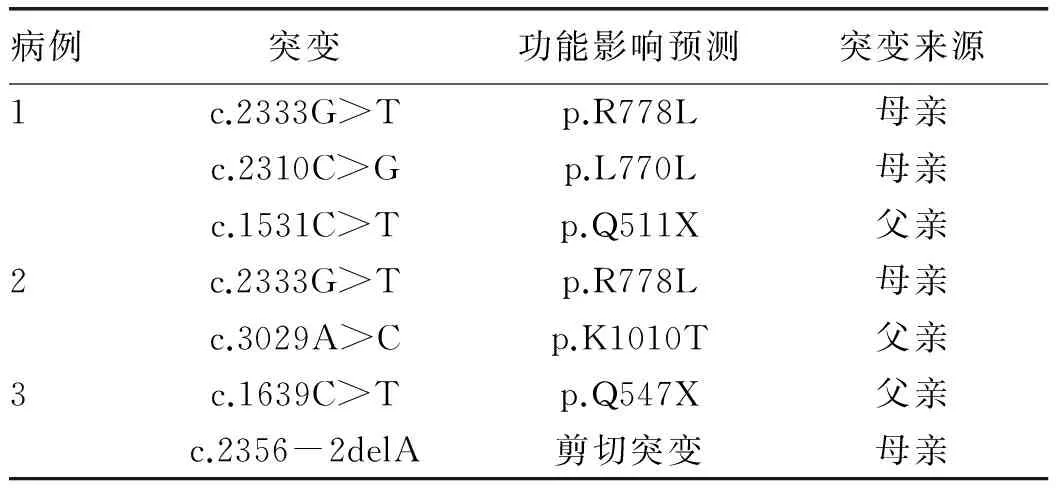
表1 ATP7B基因诊断结果
2.2 临床转归 根据我国《肝豆状核变性诊断与治疗指南》[12]及《肝衰竭诊治指南(2012年版)》[13],3例患儿均可临床诊断为“肝豆状核变性,肝硬化,肝功能失代偿”。
病例1治疗20 d后自觉双踝关节疼痛较前缓解并逐渐消失,凝血功能和低蛋白血症改善,好转出院。治疗后2个月门诊随访凝血功能和Alb明显改善(表2)。治疗前肝功能Child-Pugh评分为8分,治疗后为8分。
病例2治疗2周余血浆Alb水平即明显上升,约3个月血浆Alb水平恢复正常,约6个月随访血浆Alb水平仍正常,但凝血功能改善缓慢(表2)。治疗前肝功能Child-Pugh评分为9分,治疗后为7分。
病例3在入院初期曾输注新鲜冰冻血浆1次,在青霉胺联合葡萄糖酸锌治疗约2周后,凝血功能改善;约1个月后,凝血功能和Alb均明显好转;治疗约3个月门诊随访,凝血功能几乎恢复正常,血浆Alb水平继续改善(表2)。治疗前肝功能Child-Pugh评分为11分,治疗后为6分。
除病例1治疗初期曾因发热、WBC及PLT的减低、凝血异常加重,青霉胺暂时减量,并使用血浆改善凝血功能外,其余病例无明显不良反应,耐受性良好。动态监测血常规示WBC及PLT无明显下降反而有改善(病例1经治疗62 d后WBC与PLT分别为:3.92×109/L、55×109/L;病例2经治疗161 d后WBC与PLT分别为2.24×109/L、29×109/L;病例3经治疗81 d后WBC与PLT分别为:7.14×109/L、170×109/L)。且病例2与病例3的Child-Pugh评分均有所降低。
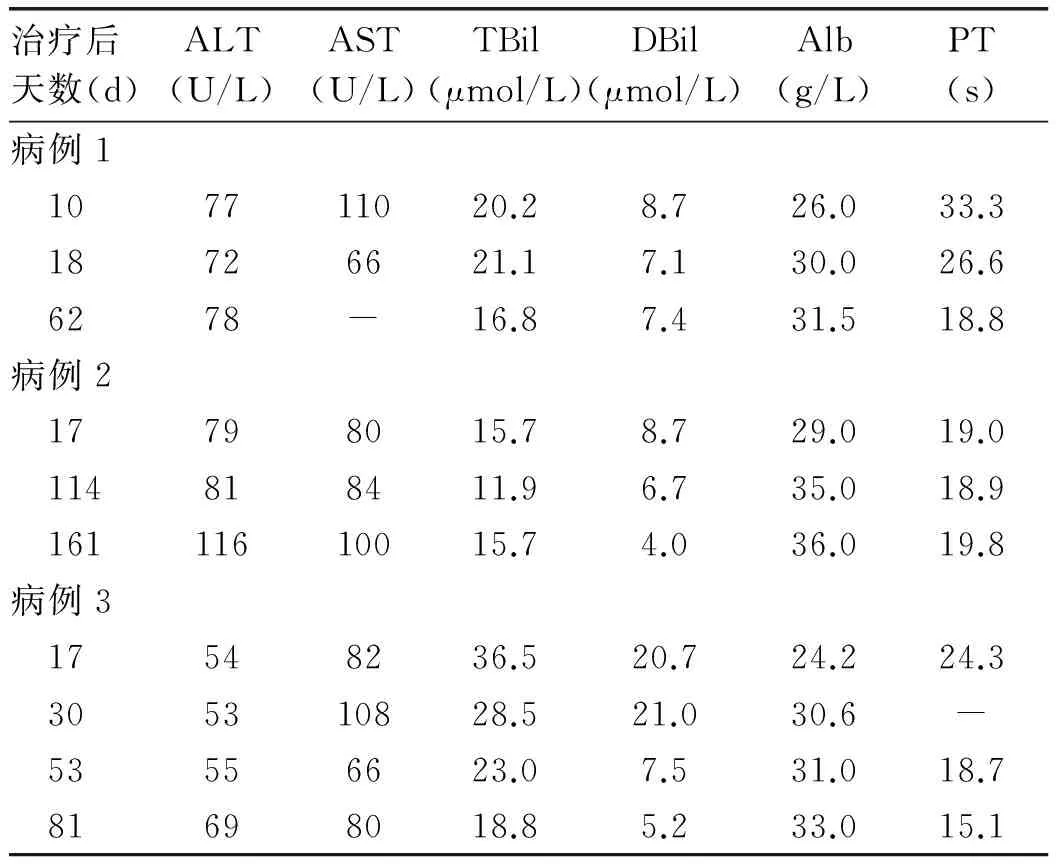
表2 治疗和随访期间肝功能指标及PT变化情况
注:ALT正常值范围7~40 U/L;AST正常值范围13~35 U/L;TBil正常值范围5~21 μmol/L;DBil正常值范围0~3.4 μmol/L;Alb正常值范围35~52 g/L;PT正常值范围9~12.5 s;“-”代表数据缺失
3 讨论
威尔逊氏病的致病基因ATP7B位于染色体13q14.3,其编码一种铜转运P型ATP酶。目前已发现的ATP7B基因突变位点有480个,其中超过300个基因的突变已被发现与威尔逊氏病有关[14]。ATP7B基因突变导致ATP酶活性减弱或丧失,血清铜蓝蛋白合成减少以及胆道排铜障碍[15],导致铜在体内蓄积。铜是细胞功能的重要元素,但游离铜具有毒性,会产生不可逆的细胞损伤。除了ATP7B基因突变,其他因素如遗传、脂质代谢,甚至环境因素都可能促使威尔逊氏病发展[16]。
目前铜蓄积致组织损伤的潜在病理发病机制尚不明确。过去一直认为威尔逊氏病细胞损伤的特点是由于过量的铜的直接毒性作用。然而,最近的证据表明组织内铜水平的增加可能会引起一系列有害的生化反应,特别是氧化应激反应,可破坏线粒体的结构和完整性,从而导致细胞损伤[17]。特别是因铜刺激导致的脂质失调被证明能够显著促进线粒体损伤[18]。有学者[19]认为人体组织铜水平的升高可引起X连锁凋亡抑制蛋白减少,导致细胞凋亡加速。另有报道[20],威尔逊氏病患者体内出现铜沉积的同时会出现铁的积累。由于血浆铜蓝蛋白氧化酶活动减弱,血浆铜蓝蛋白水平降低,亚铁转化为三价铁的稳态被打破[21],因此考虑威尔逊氏病患者出现肝损伤的部分原因可能为铁的积累[20]。然而目前铜过剩导致组织损伤的确切机制仍不完全清楚。
威尔逊氏病是目前少数有可能治愈的遗传性疾病之一,重要的是患者应在早期就得到及时有效的诊治,并且需要终生治疗(除非接受了肝移植手术)。但由于威尔逊氏病临床表现变化较大,导致早期诊断具有一定的挑战性。其原因主要是首诊医生对该病的认识及警惕性较低。根据以往经验,诊断检查应该包括:病史、临床表现、肝功能检测、血清血浆铜蓝蛋白、24 h尿铜排泄、眼科检查、肝活组织检查测量肝实质的铜水平、肝脏成像研究、基因检测[22]。其中肝活组织检查仍然是诊断威尔逊氏病的“金标准”[23]。随着人们对疾病认识的增加和医学遗传学的进步,基因检测在威尔逊氏病患者及其家庭成员,以及患有不明原因肝脏或神经精神疾病人群中,扮演越来越重要的角色[24-25]。然而,对致病基因突变进行筛查的价格非常昂贵,不适合所有人群。早期诊断可能通过大规模筛查使患者在发生症状前得到检测。建议对新生儿血液进行血清铜蓝蛋白检测或3~6岁儿童尿液进行大规模筛查[26]。
目前威尔逊氏病患者的治疗方法包括:药物治疗、对症治疗、肝移植、低铜饮食、康复及心理治疗。肝移植是威尔逊氏病患者暴发性肝衰竭或肝硬化晚期的首选治疗手段[27]。然而,对于严重病例,即使肝豆状核变性出现严重肝功能失代偿或肝衰竭,患者仍有可能通过药物治疗恢复至代偿状态。如本研究中的3例患儿,在低铜饮食的基础上,采用螯合剂和锌剂联合治疗后,肝功能各项指标逐渐改善,避免了肝移植。其中1例治疗过程中出现WBC及PLT进一步降低,经青霉胺减量后PLT逐渐回升,再次逐步增加青霉胺剂量耐受良好。
综上所述,由于肝豆状核变性临床表现多变,起病隐袭,有些患者诊断时已经有非常严重的肝硬化,甚至肝功能失代偿表现,但此时不应放弃药物治疗。恰当的药物治疗仍有可能阻止病情发展,避免肝移植。本研究的不足之处是随访时间尚短,但预测3例患儿如果规律随访,能够耐受药物治疗,将可避免肝移植,最终单用螯合剂或锌剂长期无病生存。
[1] SCHEINBERG IH, STERNLIEB I. Wilson disease and idiopathic copper toxicosis[J]. Am J Clin Nutr, 1996, 63(5): 842s-845s.
[2] CACA K, FERENCI P, KÜHN HJ, et al. High prevalence of the H1069Q mutation in East German patients with Wilson disease: rapid detection of mutations by limited sequencing and phenotype-genotype analysis[J]. J Hepatol, 2001, 35(5): 575-581.
[3] COMPSTON A. Progressive lenticular degeneration: a familial nervous disease associated with cirrhosis of the liver, by S. A. Kinnier Wilson, (From the National Hospital, and the Laboratory of the National Hospital, Queen Square, London) Brain 1912: 34; 295-509[J]. Brain, 2009, 132(Pt 8): 1997-2001.
[4] CUMINGS JN. The cerebrospinal fluid in poliomyelitis[J]. Overseas Postgrad Med J, 1949, 3(10): 163.
[5] DEHGHANI SM, IMANIEH MH, HAGHIGHAT M, et al. Etiology and complications of liver cirrhosis in children: report of a single center from southern iran[J]. Middle East J Dig Dis, 2013, 5(1): 41-46.
[6] THOMAS GR, FORBES JR, ROBERTS EA, et al. The Wilson disease gene: spectrum of mutations and their consequences[J]. Nat Genet, 1995, 9(2): 210-217.
[7] ZALI N, MOHEBBI SR, ESTEGHAMAT S, et al. Prevalence of ATP7B gene mutations in Iranian patients with Wilson disease[J]. Hepat Mon, 2011, 11(11): 890-894.
[8] YANG J, CHAN P. Gene symbol: ATP7B. Disease: Wilson′s disease[J]. Hum Genet, 2005, 118(3-4): 544.
[9] SQUITTI R, SIOTTO M, BUCOSSI S, et al. In silico investigation of the ATP7B gene: insights from functional prediction of non-synonymous substitution to protein structure[J]. Biometals, 2014, 27(1): 53-64.
[10] SCHUSHAN M, BHATTACHARJEE A, BEN-TAL N, et al. A structural model of the copper ATPase ATP7B to facilitate analysis of Wilson disease-causing mutations and studies of the transport mechanism[J]. 2012, 4(7): 669-678.
[11] ZHU M, NI W, DONG Y, et al. EGFP tags affect cellular localization of ATP7B mutants[J]. CNS Neurosci Ther, 2013,19(5): 346-351.
[12] Professional Study Group on Parkinson′s Disease and Movement Disorders, Professional Study Group on Neurogenetic Diseases, Neurology Branch of Chinese Medical Association. Guidelines for the diagnosis and treatment of hepatolenticular degeneration[C]//Neurology Branch of Chinese Medical Association. Collected Papers from the 11th National Conference of Neurology. Changchun, 2008: 5. (in Chinese) 中华医学会神经病学分会帕金森病及运动障碍疾病专业学组、神经遗传疾病专业学组. 肝豆状核变性诊断与治疗指南[C]//中华医学会神经病学分会. 第十一届全国神经病学学术会议论文汇编. 长春, 2008: 5.
[13] Liver Failure and Artificial Liver Group, Chinese Society of Infectious Diseases, CMA; Severe Liver Diseases and Artificial Liver Group, Chinese Society of Hepatology, CMA. Guideline for diagnosis and treatment of liver failure (2012 version)[J]. Chin J Clin Infect Dis, 2012, 5(6): 321-327. (in Chinese)
中华医学会感染病学分会肝衰竭与人工肝学组, 中华医学会肝病学分会重型肝病与人工肝学组. 肝衰竭诊治指南(2012年版)[J]. 中华临床感染病杂志, 2012, 5(6): 321-327.
[14] KUCINSKAS L, JEROCH J, VITKAUSKIENE A, et al. High frequency of the c.3207C>A (p.H1069Q) mutation in ATP7B gene of Lithuanian patients with hepatic presentation of Wilson′s disease[J]. World J Gastroenterol, 2008, 14(38): 5876-5879.
[15] DONG QY, WU ZY. Advance in the pathogenesis and treatment of Wilson disease[J]. Transl Neurodegener, 2012, 1(1): 23.
[16] WU F, WANG J, PU C, et al. Wilson′s disease: a comprehensive review of the molecular mechanisms[J]. Int J Mol Sci, 2015, 16(3): 6419-6431.
[17] ROBERTS EA, ROBINSON BH, YANG S. Mitochondrial structure and function in the untreated Jackson toxic milk (tx-j) mouse, a model for Wilson disease[J]. Mol Genet Metab, 2008, 93(1): 54-65.
[18] SAUER SW, MERLE U, OPP S, et al. Severe dysfunction of respiratory chain and cholesterol metabolism in Atp7b(-/-) mice as a model for Wilson disease[J]. Biochim Biophys Acta, 2011, 1812(12): 1607-1615.
[19] MUFTI AR, BURSTEIN E, CSOMOS RA, et al. XIAP is a copper binding protein deregulated in Wilson′s disease and other copper toxicosis disorders[J]. Mol Cell, 2006, 21(6): 775-785.
[20] HAYASHI H, YANO M, FUJITA Y, et al. Compound overload of copper and iron in patients with Wilson′s disease[J]. Med Mol Morphol, 2006, 39(3): 121-126.
[21] MERLE U, TUMA S, HERRMANN T, et al. Evidence for a critical role of ceruloplasmin oxidase activity in iron metabolism of Wilson disease gene knockout mice[J]. J Gastroenterol Hepatol, 2010, 25(6): 1144-1150.
[22] European Association for Study of Liver. EASL Clinical Practice Guidelines: Wilson′s disease[J]. J Hepatol, 2012, 56(3): 671-685.
[23] BURKHEAD JL, GRAY LW, LUTSENKO S. Systems biology approach to Wilson′s disease[J]. Biometals, 2011, 24(3): 455-466.
[24] BENNETT J, HAHN SH. Clinical molecular diagnosis of Wilson disease[J]. Semin Liver Dis, 2011, 31(3): 233-238.
[25] BENNETT JT, SCHWARZ KB, SWANSON PD, et al. An exceptional family with three consecutive generations affected by Wilson disease[J]. JIMD Rep, 2013, 10: 1-4.
[26] NAKAYAMA K, KUBOTA M, KATOH Y, et al. Early and presymptomatic detection of Wilson′s disease at the mandatory 3-year-old medical health care examination in Hokkaido Prefecture with the use of a novel automated urinary ceruloplasmin assay[J]. Mol Genet Metab, 2008, 94(3): 363-367.
[27] ALA A, BORJIGIN J, ROCHWARGER A, et al. Wilson disease in septuagenarian siblings: raising the bar for diagnosis[J]. Hepatology, 2005, 41(3): 668-670.
引证本文:LI CX, GONG JY, ZHANG MH, et al. Effect of pharmacotherapy on decompensated liver function in children with hepatolenticular degeneration[J]. J Clin Hepatol, 2017, 33(8): 1543-1547. (in Chinese) 李春霞, 龚敬宇, 张梅虹, 等. 药物治疗儿童肝豆状核变性肝功能失代偿的效果观察[J]. 临床肝胆病杂志, 2017, 33(8): 1543-1547.
(本文编辑:葛 俊)
Effect of pharmacotherapy on decompensated liver function in children with hepatolenticular degeneration
LIChunxia,GONGJingyu,ZHANGMeihong,etal.
(DepartmentofInfectiousDiseases,Yan′anUniversityAffiliatedHospital,Yan′an,Shaanxi716000,China)
Objective To investigate the effect of pharmacotherapy on decompensated liver function in children with hepatolenticular degeneration. Methods Three children with “liver cirrhosis and decompensated liver function” as the onset of hepatolenticular degeneration who were admitted to Jinshan Hospital Affiliated to Fudan University from August 1, 2015 to August 1, 2016 were enrolled, and ATP7B gene sequencing was performed to make a confirmed diagnosis. Clinical outcome was recorded. Results Compound heterozygous mutation of the ATP78 gene was detected in the three children. After comprehensive medical treatment, all children had improvements in hypoproteinemia and coagulation function, and two children had a reduced Child-Pugh score. Conclusion Hepatolenticular degeneration should be considered for children with unexplained abnormal liver function and even liver cirrhosis, and early diagnosis and timely pharmacotherapy can reduce the need for liver transplantation.
hepatolenticular degeneration; liver cirrhosis; drug therapy; child
10.3969/j.issn.1001-5256.2017.08.027
2017-01-13;
2017-04-05。
李春霞(1983-),女,主治医师,主要从事慢性肝病防治研究;龚敬宇(1974-),女,副主任医师,主要从事儿童肝病及儿童呼吸道疾病的诊断和治疗研究。二者对本文贡献相同,同为第一作者。
王建设,电子信箱:jshwang@shmu.edu.cn。
R575
A
1001-5256(2017)08-1543-05
