抗生素使用对冬春季婴幼儿肠道菌群内部相关性的影响
2017-08-28何国斌李文浩虞荣斌朱中海李文静曾令霞
何国斌,孙 超,李文浩,虞荣斌,朱中海,李文静,韩 蓓,程 悦,,曾令霞
(1.西安交通大学医学部公共卫生学院流行病与卫生统计学系,陕西 西安 710061;2.陕西省营养与食品安全工程研究中心,陕西 西安 710061)
抗生素使用对冬春季婴幼儿肠道菌群内部相关性的影响
何国斌1,孙 超1,李文浩1,虞荣斌1,朱中海1,李文静1,韩 蓓2,程 悦1,2,曾令霞1
(1.西安交通大学医学部公共卫生学院流行病与卫生统计学系,陕西 西安 710061;2.陕西省营养与食品安全工程研究中心,陕西 西安 710061)
目的 研究抗生素使用对冬春季婴幼儿肠道菌群内部相关性的影响。方法 采用1:1病例对照研究设计,选取于陕西省商洛市中心医院就诊患腹泻疾病的2岁以下患儿为病例组,同期到该医院进行健康体检或免疫接种的健康婴幼儿作为对照组。采用二代测序的技术检测婴幼儿样本粪便中微生物群落的组成结构和多样性。绘制细菌属水平相关性网络图,分析各菌属之间的相关性。结果 本研究共收集粪便样本120例,最终纳入分析的样本数为115。在对照组中,拟杆菌属与其他菌属存在最多关联,大肠埃希菌/志贺菌属、不动杆菌属、芽孢杆菌属和Lysinibacillus菌属存在明显正相关性(r值分别为0.640、0.945、0.892,均P<0.05),后3种菌属数量上的增加直接影响了拟杆菌数量的减少,但拟杆菌却未表现出与大肠埃希菌/志贺菌的负相关性(r=-0.252,P>0.05);在腹泻未使用抗生素组的婴幼儿中,Akkermansia菌与其他菌属存在最多的关联,它与普雷沃氏菌属、Barnesiella菌、地杆菌存在两两正相关性(r值分别为0.735、0.798、0.833,均P<0.05),Akkermansia菌通过与芽孢杆菌和不动杆菌的正相关性(r值分别为0.473、0.401,均P<0.05),影响大肠埃希菌和Lysinibacillus菌属的数量,但Akkermansia菌、芽孢杆菌属却与大肠埃希菌没有直接的相关关系;在腹泻使用抗生素组中,地杆菌属与其他菌属联系最多,并且出现了两大正相关集团,一是以地杆菌为中心,表现为地杆菌与不动杆菌、普雷沃氏菌、Barnesiella菌、Lysinibacillus菌和芽孢杆菌属的正相关关系(r值分别为0.622、0.876、0.911、0.435、0.606,均P<0.05),另一个是以韦荣氏菌为中心,表现为韦荣氏菌与Akkermansia菌、嗜血菌和Megasphaera菌属存在正相关关系(r值分别为0.414、0.773、0.567,均P<0.05),两大集团存在拮抗作用,表现为韦荣氏菌和嗜血菌对地杆菌存在负相关(r=-0.538、-0.380,P<0.05),并且韦荣氏菌丰度的增加会抑制芽孢杆菌、普雷沃氏菌、地杆菌和Barnesiella菌属的丰度值,同时肠球菌丰度的增加也会使第一集团菌属丰度相应减少,双歧杆菌仅对芽孢杆菌存在负相关作用(r=-0.403,P<0.05)。抗生素的使用增加了肠道菌群在属水平的内部相关性复杂程度,并改变了与其他菌属联系最多的菌属类别。结论 抗生素的使用导致了婴幼儿肠道菌群微生态失衡。
肠道菌群;抗生素;婴幼儿;内部相关性
人体内定植的微生物群与人类的疾病密切相关,对人体健康有重要影响[1]。现有研究表明,婴幼儿肠道菌群的发育程度和稳定性受到多种因素影响,包括早产、分娩方式、喂养方式、抗生素和益生菌的使用等[2]。抗生素是目前最常见的儿童处方药物,而婴幼儿期是肠道菌群形成和稳定的关键时期,抗生素的使用会改变肠道菌群的组成结构和多样性,引起菌群微生态失衡,对婴幼儿的健康成长造成长远的不良影响,有关流行病学资料甚至表明许多成年疾病与早期抗生素使用相关[3]。然而,国内有关抗生素使用对婴幼儿肠道菌群影响作用的研究鲜有报道,且已有研究存在实验室检测手段落后,样本量偏少等问题。本研究利用高通量测序技术对婴幼儿的肠道菌群进行了16S rDNA基因测序,在前期探讨抗生素使用对婴幼儿肠道菌群多样性影响的基础上,进一步探讨婴幼儿期抗生素的使用对肠道菌群内部相关性的影响。
1对象和方法
1.1研究设计和对象
采用1:1病例对照研究设计。研究对象为2013年12月至2014年1月来商洛市中心医院就诊的患腹泻疾病的2岁以下患儿为病例组,同期到该医院进行健康体检或免疫接种的健康婴幼儿作为对照组。考虑到婴幼儿的健康状态可能会影响其抗生素的使用,故将全部研究对象分为3组,即对照组、腹泻未使用抗生素组和腹泻使用抗生素组。
1.2研究对象纳入标准
健康婴幼儿纳入标准:近一月内未患腹泻及其他相关疾病,无出生缺陷。腹泻婴幼儿纳入标准:符合《婴幼儿疾病诊疗指南》[4]中腹泻诊断的婴幼儿,粪便性状改变;排便次数≥3次/天或比平日增多超过1倍以上。
1.3样本量的确定
正常2岁以下婴幼儿腹泻发病率为P0=30%[5],而抗生素相关性腹泻发病率为P1=5%[6],按照95%的置信度、90%的检验效能及病例对照研究设计,则病例组和对照组各需47例,按照约20%的拒绝率,研究样本扩大至120例(每组各60例)。为确保研究样本的代表性,实际样本采集中按照婴幼儿月龄分为三层:“0~5月龄”、“6~11月龄”、“12~24月龄”,每层内分别采集20例病例及20例对照。
1.4粪便样本采集
采用一次性无菌离心管由医院指定医生采集健康及腹泻婴幼儿肠道粪便(每人份2~5g),后冻存于-80℃冰箱。为避免采集过程中粪便遭受污染,所有粪便收集后即刻用75%酒精浸泡灭菌的蜡纸封闭保存。粪便样本的转移均置于-20℃的冰盒中。
1.5细菌DNA的提取和验证
购买QIAGEN QIAamp MiniStool Kit(QIAGEN,Hilden,Germany)专用试剂盒提取粪便中的总DNA,提取出的总DNA采用NanoPhotometer TM(IMPLEN,Germany)仪器进行在260nm和280nm吸光度的检测,用260nm/280nm的吸光度比值检测提取物中总DNA的浓度,结果发现其比值均在1.7~1.9,证明纯度良好可以进行下一步实验。合成细菌16S rDNA V3~V4区片段通用引物(由上海生工生物技术服务有限公司合成),引物对为F338/R534[7],具体序列为F338(5′ACTCCTACGGGAGGCAGCAG-3′)和R534(5′ATTACCGCG ̄GCTGCTGG-3′),PCR扩增提取的总DNA,对PCR产物进行琼脂糖凝胶电泳分析,以验证细菌总DNA。
1.6高通量测序
采用Illumina公司的Miseq 250PE测序平台,对细菌16S rDNA基因V4区进行高通量测序,并对测序结果进行生物信息学分析。该过程委托深圳市千年盛世基因科技有限公司。
1.7质量控制
调查员均经过统一培训,现场严格按照标准收取样本;标准化无污染操作采集粪便,控制粪便的空气暴露时间,及时冻存粪便样本;除二代测序以外的实验操作均在标准化无污染的实验室中完成,保证粪便不引入环境中的杂菌。
1.8研究经费来源及伦理学问题
本研究数据来自于国家自然科学基金(编号:81373019)项目。并通过“西安交通大学医学院医学生物科研伦理学委员会”审批(伦理学批件号:2013-093)。所有调查包括粪便样品采集均获得婴幼儿家长的知情同意。
1.9统计学方法
流行病学资料的数据采用EPI Data 3.1软件数据录入,使用SPSS 20.0软件进行数据整理和分析。计数资料用频数、百分比表示,组间差异性比较采用χ2检验。实验室的婴幼儿肠道菌群信息采用CASAVA v1.8.2和FastQC软件,按Illumina 标准流程进行base calling及数据产出统计,用以检验本次实验精度;按标准原则对数据进行过滤,reads双端merge,按照barcode序列区分样本序列,去除barcode及引物序列,分别对每个样本和所有样本的数据产出和reads长度分布进行统计,得到各样本可操作分类单元(operational taxonomic unit,OTU)数。将检出的序列和Greengenes数据库比对进行样本物种的确定,计算划分为特定物种的OTU数目与各样本内总的OTU数目的比值,称为该物种的相对丰度。再利用各菌的相对丰度数据进行各菌种之间的相关性分析。假设检验均采用双侧检验,检验水准为0.05。
2结果
2.1一般资料
本研究共收集粪便样本120例,其中有5例经筛选后排除,最终纳入分析的样本数为115,健康对照组59例、腹泻未使用抗生素组28例和腹泻使用抗生素组28例。对婴幼儿的月龄分布进行了组间比较后发现,3组间差异无统计学意义(χ2=1.932,P=0.748),排除了月龄因素在各分组间的差异,见表1。
表1 婴幼儿月龄分布[n(%)]
Table 1 Distribution of infants’ months of age[n (%)]
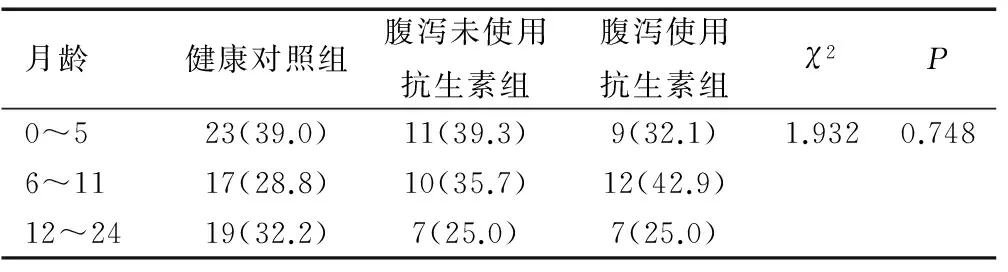
月龄健康对照组腹泻未使用抗生素组腹泻使用抗生素组χ2P0~523(39.0)11(39.3)9(32.1)1.9320.7486~1117(28.8)10(35.7)12(42.9)12~2419(32.2)7(25.0)7(25.0)
2.2菌种测序信息结果质量分析
本次进行16S rDNA V4区高通量实验测序的样本共120例,其中有5例因在提取DNA的过程中有所污染或量不足,导致测序结果失败,故最终实际参与测序的样本共115例。本次实验构建了2个shortgun文库,其中超过90%的序列碱基质量均在Q30以上,平均读长为328bp,且各文库的总序列数均超过100万条。各样本在序列接近120 000条时便已达到平台期。证明此次测序结果满足后续分析要求。
2.3肠道菌群菌种相关性分析
利用属水平占比前80%的菌属丰度信息,筛选其中15种可以明确属名称的菌属进行相关性分析(见表2~表4),并绘制重要菌属相关性网络图,描述其中具有统计学意义的菌属关系,见图1。

图1 重要菌属相关性网络图
Fig.1 Network diagram of internal correlation of main bacteria genuses
2.3.1对照组菌属相关性分析
在对照组中,与其他菌属存在最多联系关系的菌属是拟杆菌属,大肠埃希菌/志贺菌属、不动杆菌属、芽孢杆菌属和Lysinibacillus菌属存在明显正相关性(r分别为0.640、0.945、0.892,均P<0.05),后3种菌属数量上的增加直接影响了拟杆菌数量的减少,但拟杆菌却未表现出与大肠埃希菌/志贺菌的负相关性(r=-0.252,P>0.05);Barnesiella菌属与地杆菌属和普雷沃氏菌属存在正相关性(r分别为0.959、0.867,均P<0.05);拟杆菌和Akkermansia菌属存在正相关性(r=0.395,P<0.05),链球菌属与嗜血菌属也存在正相关性(r=0.329,P<0.05),见表2。

表2 对照组重点菌属相关性分析(r)
注:*按P<0.05检验水准差异有统计学意义。
2.3.2腹泻未使用抗生素组菌属相关性分析
在腹泻未使用抗生素组的婴幼儿中,Akkermansia菌与其他菌属存在最多关联,它与普雷沃氏菌属、Barnesiella菌、地杆菌存在两两正相关性(r值分别为0.735、0.798、0.833,均P<0.05),同时Akkermansia菌通过与芽孢杆菌和不动杆菌的正相关性(r值分别为0.473、0.401,均P<0.05),还可以影响大肠埃希菌和Lysinibacillus菌属的数量,但Akkermansia菌和芽孢杆菌属却与大肠埃希菌没有直接的相关关系,双歧杆菌属丰度的增加会使肠球菌和Megasphaera菌属丰度增加,后两者却无明显正相关关系;韦荣氏菌属丰度的上升直接影响了Akkermansia菌、Barnesiella菌和地杆菌属丰度的增加,但对普雷沃氏菌的影响无统计学意义,见表3。
2.3.3腹泻使用抗生素组菌属相关性分析
在腹泻使用抗生素组中,地杆菌属与其他菌属联系最多。在这组婴幼儿的肠道内出现了两大正相关集团,其一是以地杆菌为中心,表现为地杆菌与不动杆菌、普雷沃氏菌、Barnesiella菌、Lysinibacillus菌和芽孢杆菌属的明显正相关关系(r值分别为0.622、0.876、0.911、0.435、0.606,均P<0.05),另一个是以韦荣氏菌为中心,与Akkermansia菌、嗜血菌和Megasphaera菌属存在明显正相关关系(r值分别为0.414、0.773、0.567,均P<0.05),两大集团存在拮抗作用,表现为韦荣氏菌和嗜血菌与地杆菌存在负相关性(r=-0.538、-0.380,P<0.05),并且韦荣氏菌丰度的增加会抑制芽孢杆菌、普雷沃氏菌、地杆菌和Barnesiella菌属的丰度值,同时肠球菌丰度的增加也会使第一集团菌属丰度相应减少,双歧杆菌仅对芽孢杆菌存在负相关作用(r=-0.403,P<0.05),见表4。
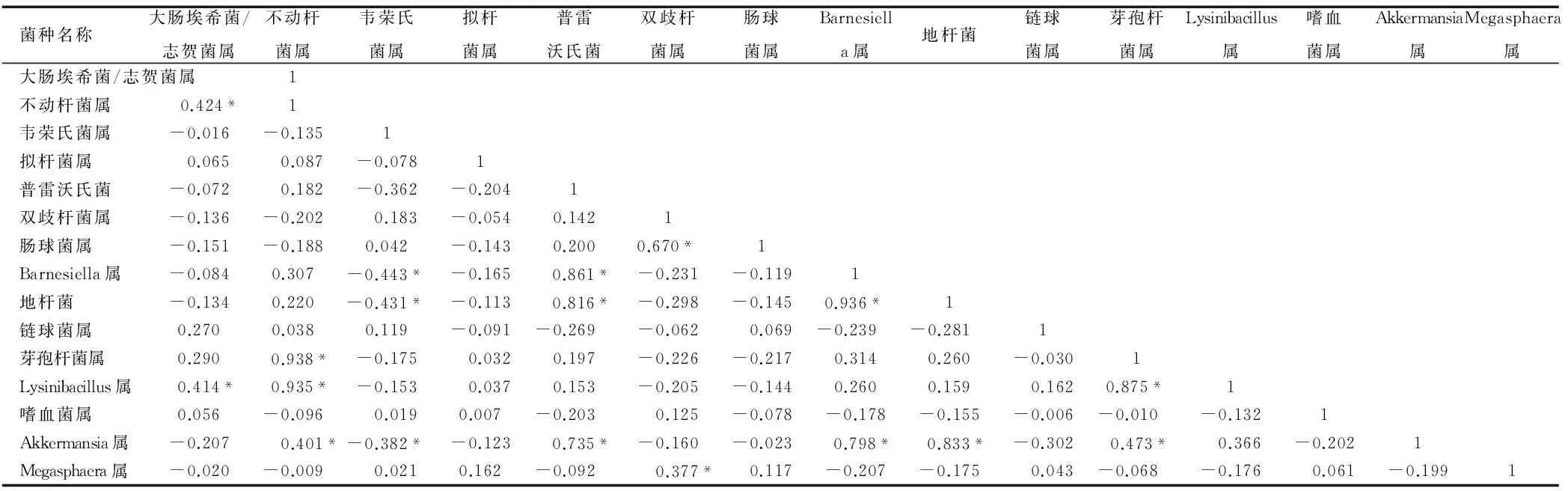
表3 腹泻未使用抗生素组重点菌属相关性分析(r)
注:*按P<0.05检验水准差异有统计学意义。
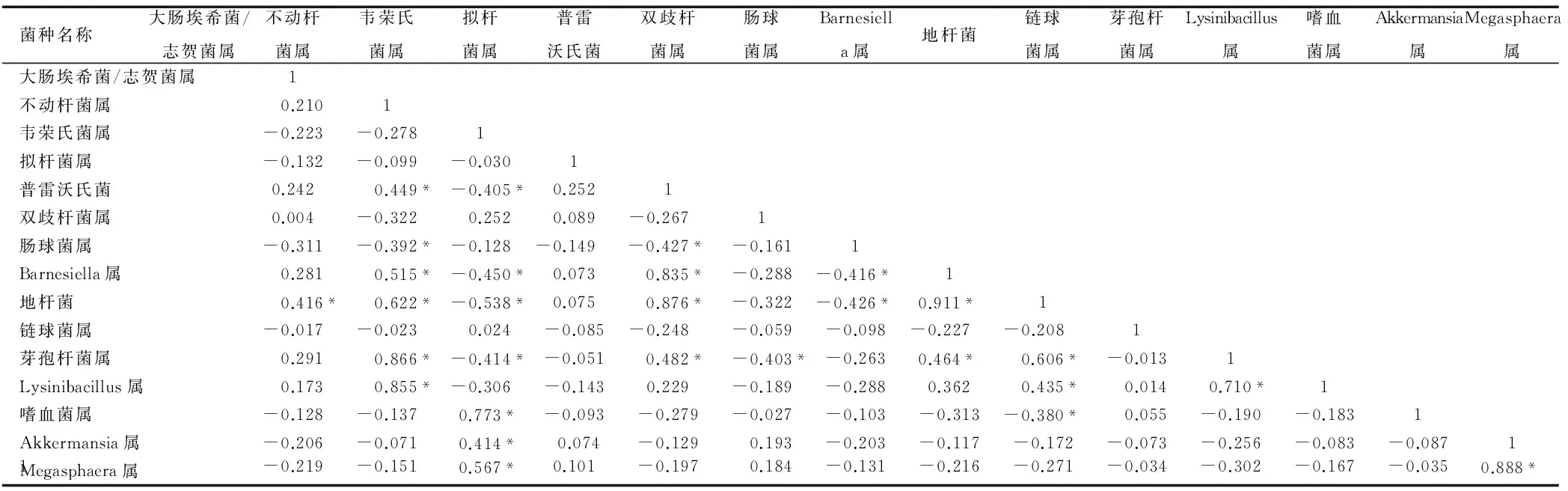
表4 腹泻使用抗生素组重点菌属相关性分析(r)
注:*按P<0.05检验水准差异有统计学意义。
3讨论
3.1本研究的主要发现
人类肠道内微生物群所表现出来的重要生理功能,如免疫调节、代谢、营养和保护作用,都是源于肠道内微生物的平衡共生体系[7]。本研究结果发现:随着抗生素的使用,肠道内菌群的相互联系变得复杂,并且起到关键作用的菌种也随之改变。说明抗生素的使用在一定程度上导致了肠道微生态失衡。而肠道内任何一种微生物功能的变异或扭曲都可能导致一系列疾病的发生,包括代谢综合征和肥胖相关疾病、肝脏疾病、肠易激综合征,炎症性肠病和结肠直肠癌等[1]。当前,许多医务人员大量使用抗生素预防手术后感染,有研究认为这不仅严重破坏了肠道菌群稳态,同时增强了抗生素耐药性的传播[8],进一步导致肠道菌群健康状况的恶化。以上均提示医疗机构要规范化抗生素使用指征,防止抗生素的滥用。
3.2腹泻和抗生素使用对肠道菌群内部相关性的影响
本研究对照组中与众细菌联系最多的菌为拟杆菌属,其直接抑制了芽孢杆菌属、Lysinibacillus菌属和不动杆菌属,并间接抑制了与上述3种菌存在正相关关系的大肠埃希菌/志贺菌属。综合考虑芽孢杆菌属、Lysinibacillus菌属、不动杆菌、大肠埃希菌/志贺菌属的致病菌属性,本研究认为拟杆菌属是健康婴幼儿人群中维持肠道菌群稳态最重要的菌属,这一结论与Palmer等于2007年的研究结果相似。随着婴幼儿由健康状态转为腹泻状态,关联最多的菌种从拟杆菌属变为Akkermansia菌属,各菌属之间的关系也变得复杂。这一结果表明腹泻和肠道菌群稳态之间有一定的关联,但两者之间的因果关系还需要进一步的研究来证实。在腹泻使用抗生素组中地杆菌属成为关联最多的菌属类型,抗生素的加入改变了原来Akkermansia菌属与韦荣氏菌的负相关关系,并且建立起了地杆菌和Lysinibacillus菌属的相关通道。说明抗生素的使用可能会改变肠道微生态环境,破坏肠道菌群定植抗力,从而促进某些致病菌的定植,这一结果与黄文献等的研究结果一致[9]。在一项最新的研究中,研究人员甚至发现抗生素通过增加营养物质的可获得性,促进了沙门菌在肠道内的茁壮成长[10]。此外,相比于腹泻未使用抗生素组,腹泻使用抗生素组的肠道微生态失调情况更加严重。这提示给婴幼儿治疗腹泻时,在不影响临床治疗效果的前提下,抗生素处方的使用应当充分考虑其对于肠道菌群的影响[11]。
3.3本研究的局限性、特点及主要结论
日常生活中婴幼儿抗生素摄入不仅来源于药品,有研究指出食物中抗生素残留也会导致肠道菌群紊乱[12],甚至即使是不同种类的抗生素摄入对肠道菌群的影响差别也非常之大[13]。由于条件所限,本研究未纳入以上因素,是局限性所在。但相比于以往实验室粪便细菌培养得到的肠道菌群数据开展分析,本研究采用Illumina二代基因测序技术,对细菌的测量和分型达到了基因水平,并利用序列拷贝数进行细菌定量,做到细菌分析的最小误差水平。同时按不同菌属分类分析相关性得到关系图网从而实现降维归类,得出的结果准确直观,能够较为准确地反映抗生素使用对肠道菌群内部相关性的影响,为规范化婴幼儿抗生素使用提供了科学依据。综上所述,抗生素的使用导致了婴幼儿肠道菌群微生态失衡,医疗机构要规范化抗生素使用指征,在不影响临床疗效的前提下,充分考虑其对肠道菌群的影响,防止抗生素滥用。
有研究发现人类肠道微生物的初始状态能在一定程度上决定抗生素对其的影响,提出监测微生物组治疗前的初始成分可以帮助预防一些抗生素或其他治疗方法带来的不利影响[14],这对于下一步的研究方向有着重要的指导意义。总之,建立一个庞大而多样化的基线队列来探索和定义婴幼儿肠道菌群的发展过程及其影响因素,探讨生命早期抗生素使用对肠道微生物结构变化的作用机制,对于预防和治疗小儿肠道微生态失衡具有十分重要的意义,同时将能在大量实验证据的基础上,为婴幼儿期抗生素的进一步规范化使用提供指导性建议。
致谢:感谢陕西省商洛市中心医院的支持和帮助,感谢所有配合调查的家庭及曾经参与过项目的学生!
[1]Marchesi J R,Adams D H,Fava F,etal.The gut microbiota and host health:a new clinical frontier[J].Gut,2016,65(2):330-339.
[2]Goulet O.Potential role of the intestinal microbiota in programming health and disease[J].Nutr Rev, 2015,73(Suppl 1):32-40.
[3]Vangay P,Ward T,Gerber J S,etal.Antibiotics,pediatric dysbiosis,and disease[J].Cell Host Microbe,2015,17(5):553-564.
[4]罗小平,刘铜林.儿科疾病诊疗指南[M].3版.北京:科学出版社,2014:134-135.
[5]Walker C L,Rudan I,Liu L,etal.Global burden of childhood pneumonia and diarrhoea.[J].Lancet,2013,381(9875):1405-1416.
[6]Varughese C A,Vakil N H,Phillips K M.Antibiotic-associated diarrhea: a refresher on causes and possible prevention with probiotics--continuing education article[J].J Pharm Pract,2013,26(5):476-482.
[8]Zaura E,Brandt B W,Teixeira de Mattos M J,etal.Same exposure but two radically different responses to antibiotics: resilience of the salivary microbiome versus long-term microbial shifts in feces[J].MBio, 2015,6(6): e01693-15.
[9]黄文献,王和平,王玉蕾,等.高通量测序检测肺炎婴幼儿抗生素治疗前后肠道菌群变化[J].中国微生态学杂志,2016,28(5):497-500.
[10]Faber F,Tran L,Byndloss M X,etal.Host-mediated sugar oxidation promotes post-antibiotic pathogen expansion[J].Nature,2016,534(7609):697-699.
[11]Korpela K,Salonen A,Virta L J,etal.Intestinal microbiome is related to lifetime antibiotic use in Finnish pre-school children[J].Nat Commun,2016,7:10410.
[12]Cerniglia C E,Pineiro S A,Kotarski S F.An update discussion on the current assessment of the safety of veterinary antimicrobial drug residues in food with regard to their impact on the human intestinal microbiome[J].Drug Test Anal,2016,8(5-6):539-548.
[13]Vibet M A,Roux J,Montassier E,etal.Systematic analysis of the relationship between antibiotic use and extended-spectrum beta-lactamase resistance in Enterobacteriaceae in a French hospital:a time series analysis[J].Eur J Clin Microbiol Infect Dis,2015,34(10):1957-1963.
[14]Raymond F,Ouameur A A,Deraspe M,etal.The initial state of the human gut microbiome determines its reshaping by antibiotics[J].ISME J,2016,10(3):707-720.
[专业责任编辑:杨文方]
Effect of antibiotics on internal correlation of intestinal microflora in infants and young children in winter and spring season
HE Guo-bin1, SUN Chao1, LI Wen-hao1, YU Rong-bin1, ZHU Zhong-hai1, LI Wen-jing1, HAN Bei2, CHENG Yue1,2, ZENG Ling-xia1
(1.Department of Epidemiology and Health Statistics, School of Public Health, Health Science Center, Xi’an Jiaotong University, Shaanxi Xi’an 710061, China; 2.Nutrition and Food Safety Engineering Research Center of Shaanxi Province, Shaanxi Xi’an 710061, China)
Objective To explore the effect of antibiotics on internal correlation of intestinal microflora in infants and young children in winter and spring season. Methods A case-control by 1:1 study design was employed in this research. Infants under 2 years old with diarrhea visiting Shangluo Central Hospital of Shaanxi Province were chosen as case group. At the same period, healthy infants coming to the same hospital for health examination or immunization injection were selected as control group. Structure and diversity of microbial communities in feces of infants and young children were detected by second generation sequencing. Bacteria genus correlation network map was drawn and internal correlations of microflora were analyzed. Results Altogether 120 stool samples were collected in this study and finally 115 samples were included in analysis. In control group, bacteroides were most correlated with other bacteria genus. Escherichia coli/Shigella, Acinetobacter, Bacillus had obvious positive correlation with Lysinibacillus (rvalue was 0.640, 0.945 and 0.892, respectively, allP<0.05). Quantity increase in latter three bacteria genuses directly caused quantity decrease in bacteroides, but bacteroides did not show negative correlation with Escherichia coli/Shigella (r=-0.252,P>0.05). In infants with diarrhea not treated with antibiotics, Akkermansia had most correlation with other bacteria genuses. It had pairwise positive correlation with Prevotella, Barnesiella and terrabacter (rvalue was 0.735, 0.798 and 0.833, respectively, allP<0.05). Through positive correlation with bacillus and Acinetobacter (rvalue was 0.473 and 0.401, respectively, bothP<0.05), Akkermansia influenced quantity of Escherichia coli and Lysinibacillus. But Akkermansia and bacillus had no direct correlation with Escherichia coli. In infants with diarrhea treated with antibiotics, terrabacter had most correlation with other bacteria genuses. Two positive correlation groups appeared. One group was centered with terrabacter, showing positive correlation of terrabacter with Acinetobacter, Prevotella, Barnesiella, Lysinibacillus and Bacillus (rvalue was 0.622, 0.876, 0.911, 0.435 and 0.606, respectively, allP<0.05). The other group was centered with Veillonella, showing positive correlation of Veillonella with Akkermansia, hemophilus and Megasphaera (rvalue was 0.414, 0.773 and 0.567, respectively, allP<0.05). There was antagonistic effect between two groups, representing by negative correlation of Veillonella andhemophilus with terrabacter (rvalue was -0.538 and -0.380, respectively, bothP<0.05). Abundance increase in Veillonella inhibited abundance of bacillus, Prevotella, terrabacter and Barnesiella. Meanwhile, abundance increase in enterococcus could cause abundance decrease in first group bacteria genuses. Bacillus bifidus only had negative correlation with bacillus (r=-0.403,P<0.05). Antibiotics usage increased the complexity of internal correlations of intestinal flora at genus level and altered bacteria genus which had most internal correlations with other bacteria. Conclusion Antibiotics usage induces dysbiosis of intestinal microflora communities in infants and young children.
intestinal microflora; antibiotics; infants and young children; internal correlation
2017-03-20
国家自然科学基金资助项目(编号:81373019)
何国斌(1993-),男,在读硕士研究生,主要从事妇幼保健及人群健康评价的研究;
孙 超(1990-),男,医师,硕士,主要从事妇幼健康及人群健康评价研究(并列第一作者)。
曾令霞,副教授/博士生导师。
10.3969/j.issn.1673-5293.2017.07.002
R725
A
1673-5293(2017)07-0753-05
