[C4Pyr]Cl/nTsOH型低共熔溶剂的制备及其氧化脱除模拟油中的二苯并噻吩
2017-08-15侯良培赵荣祥李秀萍
侯良培,赵荣祥,李秀萍
(1.辽宁石油化工大学 化学化工与环境学部 石油化工学院,辽宁 抚顺 113001;2.辽宁石油化工大学 化学化工与环境学部 化学与材料科学学院,辽宁 抚顺 113001)
[C4Pyr]Cl/nTsOH型低共熔溶剂的制备及其氧化脱除模拟油中的二苯并噻吩
侯良培1,赵荣祥1,李秀萍2
(1.辽宁石油化工大学 化学化工与环境学部 石油化工学院,辽宁 抚顺 113001;2.辽宁石油化工大学 化学化工与环境学部 化学与材料科学学院,辽宁 抚顺 113001)
通过简单加热氯代正丁基吡啶([C4Pyr]Cl)和对甲苯磺酸(TsOH)的混合物制备了[C4Pyr]Cl/nTsOH,(n = 0.1,0.2,0.3)型低共熔溶剂。以[C4Pyr]Cl/nTsOH为催化剂和萃取剂,H2O2为氧化剂组成萃取-催化氧化脱硫体系氧化脱除模拟油中的硫化物。通过FTIR表征,确定[C4Pyr]Cl/0.2TsOH的结构以及氧化产物,并考察了不同脱硫体系、n(TsOH)∶n([C4Pyr]Cl)、低共熔溶剂加入量、反应温度、n(H2O2)∶n(二苯并噻吩)和含硫化物类型对脱硫效果的影响。实验结果表明,在低共熔溶剂[C4Pyr]Cl/0.2TsOH加入量1.00 mL、反应温度50 ℃、n(H2O2)∶n(二苯并噻吩)= 6、模拟油用量5 mL的反应条件下,[C4Pyr]Cl/0.2TsOH对二苯并噻吩、4,6-二甲基二苯并噻吩和苯并噻吩的脱硫率分别达98.2%,96.0%,40.2%。由一级动力学方程和Arrhenius方程计算氧化脱除二苯并噻吩所需的表观活化能约为51.95 kJ/mol。[C4Pyr]Cl/0.2TsOH回收利用5次后,脱硫率仍不低于95.1%。
[C4Pyr]Cl/0.2TsOH;萃取-催化氧化脱硫;低共熔溶剂;表观活化能
油品中硫化物的污染给人类的生活带来严重影响。许多国家将油品中的硫含量限制在10 µg/g[1]。工业上通常用加氢脱硫(HDS)工艺脱除原油中的硫化物。但是,HDS反应条件十分苛刻(高温、高压、需大量H2),且不能有效地脱除噻吩类硫化物[2-3]。因此,以氧化、吸附、萃取、光催化和电化学为基础的非HDS工艺引起了广泛关注[3-8]。其中氧化脱硫工艺因具有反应条件温和(常温、常压),工艺简单,设备要求低,能高效地脱除噻吩类硫化物而成为研究的热点。
低共熔溶剂由氢键供体和氢键受体组成,具有易合成、原材料便宜易得以及诸多良好的物理化学性质,而且可以通过改变氢键受体和供体的化学计量比调节其性质。低共熔溶剂理论已广泛应用于很多领域,如催化、有机合成、气体吸收、材料合成等[9-12]。近年来,低共熔溶剂被应用到脱硫领域,而且很快得到了广泛关注[13-14]。但将低共熔溶剂直接用于萃取脱硫,脱硫率不高,要实现深度脱硫往往需要多级萃取才能够达到[13]。研究发现,在酸性低共熔溶剂中加入合适的氧化剂组成萃取与催化氧化相结合的新脱硫工艺极大地提高了脱硫效率。李佳慧等[14]研究了ChCl/ C2H2O4氧化脱除模拟油中的硫化物,结果表明,二苯并噻吩(DBT)的脱除率可达到95%。Liu等[15]以ChCl/2PEG为催化剂,脱硫率高达99.1%。Lü等[16]合成了TBAC/2C2H2O4,并以H2O2为氧化剂,对DBT的脱除率达到了98%。
本工作采用简单加热搅拌的方法,以氯代正丁基吡啶([C4Pyr]Cl)和对甲苯磺酸(TsOH)为原料合成了一系列[C4Pyr]Cl/nTsOH(n = 0.1,0.2,0.3)型低共熔溶剂,并将其作为催化剂和萃取剂,以H2O2为氧化剂,萃取-催化氧化脱除油品中的硫化物。系统地研究了不同反应条件对脱硫效果的影响,并探讨了脱硫反应机理。
1 实验部分
1.1 试剂和仪器
TsOH(纯度不小于98.5%(w))、4,6-二甲基二苯并噻吩(4,6-DMDBT,纯度99%(w))、DBT(纯度不小于99.0%(w))、苯并噻吩(BT,纯度97%(w)):阿拉丁试剂有限公司;H2O2(30%(w)):辽宁泉瑞试剂有限公司;CCl4(纯度99%(w))、正辛烷(纯度98%(w)):国药集团化学试剂有限公司;[C4Pyr]Cl(纯度99%(w)):上海成杰化工有限公司。
Agilent-7820型气相色谱仪:FID检测,美国安捷伦科技有限公司;NEXUS 870型傅里叶变换红外光谱仪:KBr压片,美国尼高力仪器公司;AV-400型核磁共振仪:德国Bruker公司。
1.2 低共熔溶剂[C4Pyr]Cl/nTsOH的合成
将[C4Pyr]Cl和TsOH分别按一定摩尔比(1∶0.1,1∶0.2,1∶0.3)加入到50 mL的小烧杯中混合均匀。在80 ℃下加热并搅拌至固体完全溶解为澄清、黏稠状液体,得到低共熔溶剂[C4Pyr]Cl/ nTsOH,合成反应见图1。[C4Pyr]Cl/0.2TsOH的物性参数见表1。
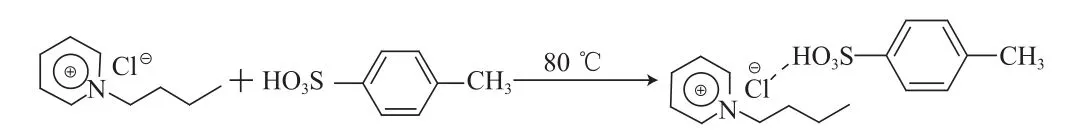
图1 [C4Pyr]Cl/nTsOH的合成Fig.1 The synthesis of[C4Pyr]Cl/nTsOH.

表1 低共熔溶剂[C4Pyr]Cl/0.2TsOH的物性参数Table 1 Physical parameters of deep eutectic solvents(DESs)[C4Pyr]Cl/0.2TsOH
1.3 催化氧化脱硫过程
分别将DBT,BT,4,6-DMDBT溶解到正辛烷中,配成3种含量均为500 µg/g的模拟油。将适量低共熔溶剂、H2O2和5 mL模拟油加入到50 mL带有冷凝装置的三角瓶中,在一定温度下进行反应。每隔20 min吸取少量上层油相,通过GC分析硫含量,并利用公式(1)计算脱硫率(η)。

式中,c0为初始硫含量,μg/g;ct为反应t时刻的硫含量,μg/g。
2 结果与讨论
2.1 低共熔溶剂的表征结果
低共熔溶剂的FTIR谱图见图2。[C4Pyr]Cl的FTIR谱图中2 959 cm-1附近的吸收峰归属于吡啶环取代基上的—CH3和—CH2的非对称伸缩振动吸收峰;1 631 cm-1和1 485 cm-1处的吸收峰归属于吡啶环的骨架振动吸收峰;1 170 cm-1和684 cm-1处的吸收峰归属于吡啶环上C—H的变形和弯曲振动吸收峰[17];771 cm-1处的吸收峰归属于吡啶环上C—C弯曲振动峰。由TsOH的FTIR谱图可见,3 424 cm-1处的吸收峰归属于—OH的伸缩振动吸收峰;1 600,1 453,1 495 cm-1处的吸收峰归属于苯环的骨架振动吸收峰;1 031 cm-1和1 006 cm-1处的吸收峰归属于S==O的伸缩振动吸收峰;680 cm-1和566 cm-1处的吸收峰分别归属于C—S的伸缩振动和SO2的剪式振动吸收峰[18]。[C4Pyr]Cl/0.2TsOH的FTIR谱图上所有的吸收峰均与[C4Pyr]Cl和TsOH一致,但相比于TsOH,3 200~3 600 cm-1间形成了较强的宽峰,上述现象表明[C4Pyr]Cl和TsOH形成了分子间氢键[19]。

图2 低共熔溶剂的FTIR谱图Fig.2 FTIR spectrum of DESs.
为进一步确定[C4Pyr]Cl和TsOH形成了分子间氢键,对合成的低共熔溶剂进行了1H NMR分析,结果见图3。由图3可知,[C4Pyr]Cl/0.2TsOH的1H NMR谱图上TsOH在δ = 6.53和[C4Pyr]Cl在δ = 4.65处的氢峰消失,而是在δ= 7.03和δ= 5.04处出现了新的氢峰。这同样证实了[C4Pyr]Cl和TsOH形成了氢键。
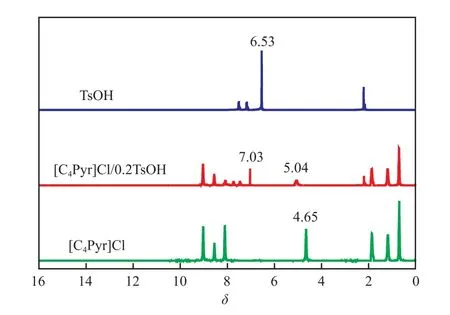
图3 低共熔溶剂的1H NMR谱图Fig.3 1H NMR spectra of DESs.
2.2 不同脱硫体系对脱硫效果的影响
不同脱硫体系对脱硫率的影响见表2。由表2可见,只有[C4Pyr]Cl和[C4Pyr]Cl/0.2TsOH的萃取作用时,脱硫率仅为10.4%和11.2%;在[C4Pyr]Cl体系中加入H2O2后,脱硫率增至12.6%;当先加入H2O2进行氧化,再用[C4Pyr]Cl/0.2TsOH作为萃取剂进行萃取时,脱硫率为10.5%;但当采用[C4Pyr]Cl/0.2TsOH作为萃取剂和催化剂,同时加入H2O2作为氧化剂形成萃取-催化氧化脱硫体系后脱硫率急剧增加至98.2%。由此可见,TsOH的引入使低共熔溶剂的催化能力得到极大提升。这是因为TsOH可以为催化氧化脱硫提供更为适宜的酸性反应环境。[C4Pyr]Cl/0.2TsOH易被H2O2氧化成活性更高的过氧化物,使脱硫效率大幅提高。因此,选择[C4Pyr]Cl/0.2TsOH氧化脱硫体系进行进一步研究。

表2 不同脱硫体系对脱硫率的影响Table 2 Influence of different desulfurization systems for removal of dibenzothiophene(DBT)
2.3 TsOH与[C4Pyr]Cl的配比对脱硫效果的影响
催化剂酸性对催化剂的活性有较大影响,因此通过改变TsOH和[C4Pyr]Cl的配比改变低共熔溶剂的酸性,进而考察其对脱硫性能的影响,结果见表3。由表3可见,当n(TsOH)∶n([C4Pyr]Cl)由0.1增至0.2时,脱硫率从95.4%升至98.2%。这是因为随TsOH的增加,低共熔溶剂的pH从1.34降至0.96,催化剂酸性增强,提高了催化剂的催化活性,氧化脱硫效率提高。继续增加TsOH的用量,体系脱硫率不仅未升高反而降至92.4%。这是因为,低共熔溶剂的酸性过强(pH = 0.52),会加剧H2O2的分解,使氧化效率降低[20]。所以,以下实验均以[C4Pyr]Cl/0.2TsOH为催化剂和萃取剂。
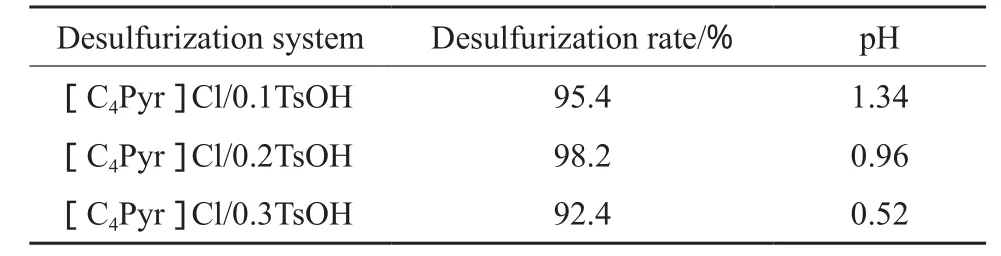
表3 n(TsOH)∶n([C4Pyr]Cl)对脱硫效果的影响Table 3 The influence of n(TsOH)∶n([C4Pyr]Cl) on desulfurization
2.4 反应温度对脱硫效果的影响
反应温度是影响脱硫效率的重要因素之一。反应温度对脱硫率的影响见图4。
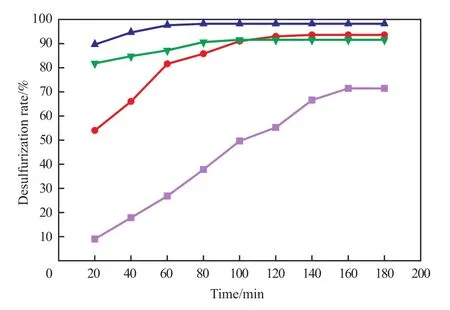
图4 反应温度对脱硫率的影响Fig.4 The influence of temperature on desulfurization.
由图4可见,随温度的升高脱硫率迅速增加;当温度为30 ℃时,脱硫率仅为71.4%;当温度升至40 ℃和50 ℃时,脱硫率显著增加到93.6%和98.2%,对应的反应平衡时间也从160 min降低到了140 min和80 min;继续升高温度至60 ℃,脱硫率降至91.6%。升高反应温度能够提高催化剂的催化活性,也能使更多的基态分子吸收能量转化为活化分子,从而增加反应体系中活化分子的浓度,使氧化效率增加[21];同时反应温度的升高,还增加了低共熔溶剂的流动性,加强了传质过程,使催化剂和DBT接触的机会增多。但反应温度过高使H2O2的分解速率加快,氧化剂的利用率下降[16]。因此,选择50 ℃为最佳的反应温度。
2.5 氧化剂用量对脱硫效果的影响
氧化剂用量直接影响反应体系中过氧化物的浓度。n(H2O2)∶n(DBT)对脱硫率的影响见图5。由图5可见,当n(H2O2)∶n(DBT)= 0时,只发生了萃取反应,脱硫率仅为12.0%。随着氧化剂用量的增加,反应体系内的过氧化物浓度大幅增加,脱硫率明显增加[22]。当n(H2O2)∶n(DBT)由2增至6时,脱硫率从90.0%增至98.2%,反应平衡时间从120 min缩短至80 min。继续增加n(H2O2)∶n(DBT)至8时,脱硫率下降到96.4%。在氧化剂增加的同时有更多的水加入到了反应体系中,对低共熔溶剂起到了稀释作用,影响了其萃取能力,阻碍了脱硫反应的进程[23]。因此,选择n(H2O2)∶n(DBT)= 6较适宜。
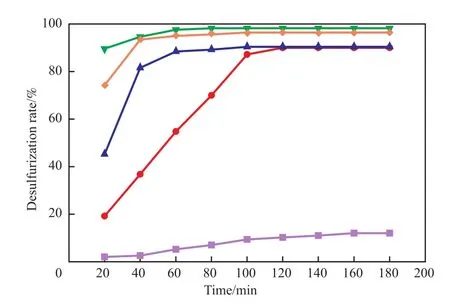
图5 n(H2O2)∶n(DBT)对脱硫率的影响Fig.5 The influence of n(H2O2)∶n(DBT) on desulfurization.
2.6[C4Pyr]Cl/0.2TsOH加入量对脱硫效果的影响
[C4Pyr]Cl/0.2TsOH加入量对脱硫率的影响见图6。由图6可见,当[C4Pyr]Cl/0.2TsOH加入量由0.50 mL增至1.00 mL时,脱硫率从96.0%升至98.2%。[C4Pyr]Cl/0.2TsOH既作为催化剂,又作为萃取剂,增加[C4Pyr]Cl/0.2TsOH的用量,同时可提高它的萃取能力,单位时间内可从模拟油中萃取出更多的DBT分子,使催化剂和DBT分子的接触机会增加,脱硫效率提高[24]。当继续增加[C4Pyr]Cl/0.2TsOH用量至1.25 mL,脱硫率基本不再升高。因此,选择适宜的[C4Pyr]Cl/0.2TsOH加入量为1.00 mL。
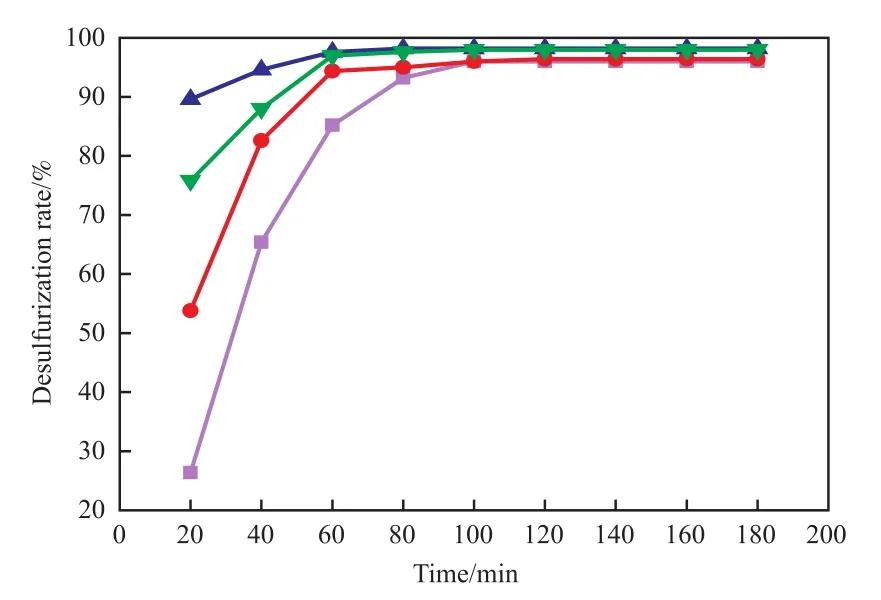
图6 [C4Pyr]Cl/0.2TsOH加入量对脱硫率的影响Fig.6 The influence of[C4Pyr]Cl/0.2TsOH amount on desulfurization.
2.7 不同硫化物的脱除和动力学分析
在本实验中,低共熔溶剂[C4Pyr]Cl/0.2TsOH对DBT的脱除效率很高,但考虑到实际油品中含硫化合物的多样性,进一步研究了其对4,6-DMDBT和BT的脱除情况,实验结果见图7。由图7a可见,[C4Pyr]Cl/0.2TsOH对DBT,4,6-DMDBT,BT的脱硫率分别达到了98.2%,96.0%,40.2%。催化剂很容易脱除DBT和4,6-DMDBT,但较难脱除BT。这主要与硫化物上S原子的电子云密度有关。电子云密度越大,S原子的活性越高,硫化物越容易被氧化脱除[16]。DBT,4,6-DMDBT,BT对应的电子云密度分别为5.758,5.760,5.739[25]。但实验结果表明,催化剂对4,6-DMDBT的脱除效果不如DBT。这是由于4,6-DMDBT苯环上的两个甲基使空间位阻增加,S原子的活性降低,氧化难度增加[26]。
为进一步验证上述结论,对不同硫化物的脱除进行动力学分析。研究表明,氧化脱硫反应符合伪一级动力学[27]。因此,可用一级动力学方程(式(2))对不同硫化物进行动力学分析。

式中,k为反应速率常数,min-1;t为反应时间,min。
由图7b可见,3条曲线的相关系数均大于0.95,说明线性关系良好,符合理论要求。以DBT,4,6-DMDBT,BT为模拟油时的反应速率常数分别 为0.030 36,0.029 28,0.003 16 min-1, 即kDBT>k4,6-DMDBT> kBT。反应速率常数越大,反应速率越快。因此,动力学模拟的结果显示[C4Pyr]Cl/0.2TsOH对不同硫化物的脱除顺序为DBT > 4,6-DMDBT >BT。这与实际实验结果一致。

Fig.7 不同硫化物的脱除和动力学分析Fig.7 The removal and kinetic analysis for different sulfides.
2.8 氧化脱除DBT的表观活化能
活化能的高低表明反应进行的难易程度。通过一级动力学方程和Arrhenius方程,对氧化脱除DBT的活化能进行模拟计算[28],结果见图8。由图8a可知,当反应温度分别为30,40,50 ℃时,反应速率常数分别为0.008 51,0.019 51,0.030 36。根据Arrhenius方程(式(3))估算出氧化脱除DBT所需的表观活化能(Ea)。

式中,R为摩尔气体常数,kJ/(mol·K);T为热力学温度,K;A为指数前因子,min-1。
由Arrhenius方程可知,通过lnk和1/T可求得表观活化能。由图8b可见,在该反应体系下氧化脱除DBT的表观活化能约为51.95 kJ/mol。低于Li等[29-30]所报道的56 kJ/mol和66.5 kJ/mol,表明以低共熔溶剂为催化剂和萃取剂、H2O2为氧化剂的氧化脱硫反应较易发生。

图8 脱除DBT的表观活化能Fig.8 The apparent activation energy(Ea) for the removal of DBT.
2.9[C4Pyr]Cl/0.2TsOH的回收利用
反应结束后,采用倾倒法回收[C4Pyr]Cl/ 0.2TsOH。将回收的[C4Pyr]Cl/0.2TsOH用等量的CCl4反复萃取4次,并用旋转蒸发仪蒸出残留溶剂,然后放入真空干燥箱中于70 ℃下干燥24 h,并利用1H NMR对回收到的[C4Pyr]Cl/0.2TsOH进行表征,结果见图9。由图9可知,反应前后试样的1H NMR谱图基本一致,确定回收到的低共熔溶剂为[C4Pyr]Cl/0.2TsOH。回收的[C4Pyr]Cl/0.2TsOH的1H NMR谱图中出现了正辛烷的特征峰(δ = 0.92,1.32),这是由于回收时少量的正辛烷未除尽。
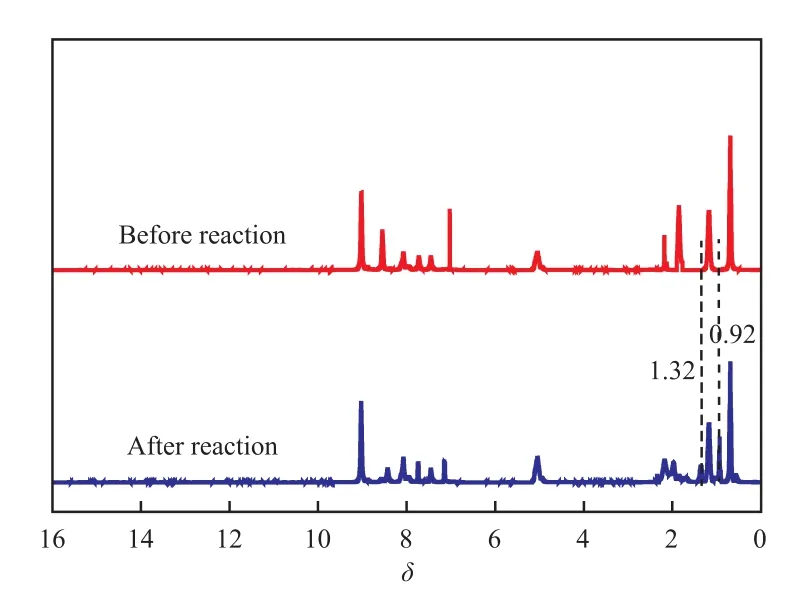
图9 反应前后[C4Pyr]Cl/0.2TsOH的1H NMR谱图Fig.9 1H NMR spectra of[C4Pyr]Cl/0.2TsOH before and after reaction.
在最佳反应条件下,向回收的[C4Pyr]Cl/ 0.2TsOH中加入新鲜的H2O2和DBT继续进行反应,[C4Pyr]Cl/0.2TsOH的回收利用情况见图10。由图10可见,[C4Pyr]Cl/0.2TsOH循环利用5次后,脱硫率略微下降,但依然高达95.1%。脱硫率下降主要是由于在反萃取过程中有少量的硫化物无法被完全萃取出来而逐渐累积造成的[31],且在回收过程中也可能会有少量的[C4Pyr]Cl/0.2TsOH损耗。

图10 [C4Pyr]Cl/0.2TsOH的回收利用情况Fig.10 The recycling of[C4Pyr]Cl/0.2TsOH. Reaction conditions referred to Table 2.
2.10 氧化脱硫机理
采用CCl4反萃取反应后[C4Pyr]Cl/0.2TsOH相中的硫化物,然后采用旋转蒸发仪除掉CCl4,得到白色的结晶,对其进行FTIR分析,结果见图11。在1 286,1 165,1 047 cm-1处的吸收峰是二苯并噻吩砜(DBTO2)和二苯并噻吩亚砜(DBTO)的特征吸收峰[32]。因此可以确定在本实验中DBT被氧化成DBTO2和DBTO。H2O2能将酸性离子液体中的含氧酸氧化成活性更高的过氧化物[1,33-34]。本实验中,认为H2O2将[C4Pyr]Cl/0.2TsOH中的磺酸基氧化成相应的过氧化态。因此,提出如图12所示的氧化脱硫机理。首先,[C4Pyr]Cl/0.2TsOH具有极强的极性,吡啶阳离子和DBT都具有芳香结构。吡啶环上的π键和DBT上的芳香结构形成π-π共轭效应,从而将模拟油中的硫化物萃取到低共熔溶剂相。在低共熔溶剂相中H2O2将—SO3H氧化成相应的—SO3H[O],然后DBT被—SO3H[O]氧化成对应的亚砜或砜。反应体系的萃取平衡被打破,低共熔溶剂继续从模拟油中萃取硫化物,直到H2O2被完全消耗或硫化物被完全氧化。
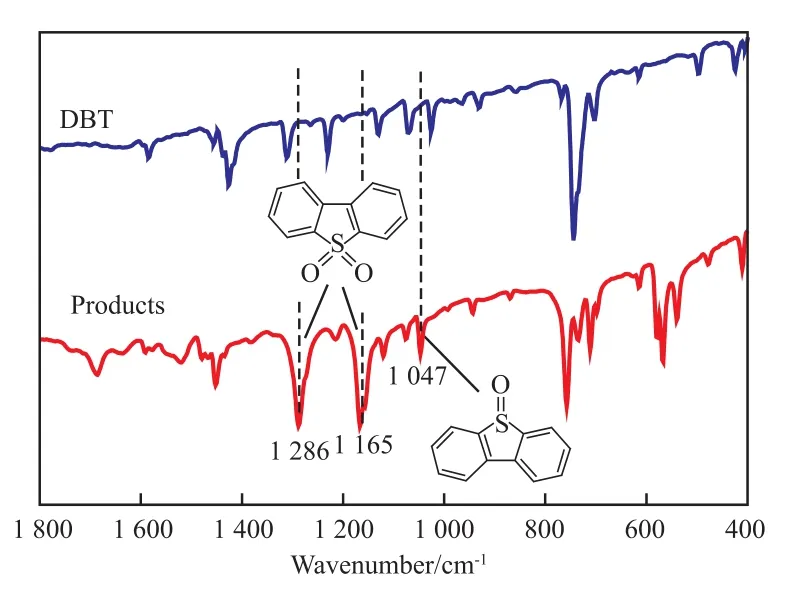
图11 氧化产物的FTIR谱图Fig.11 FTIR spectrum of oxidative products.
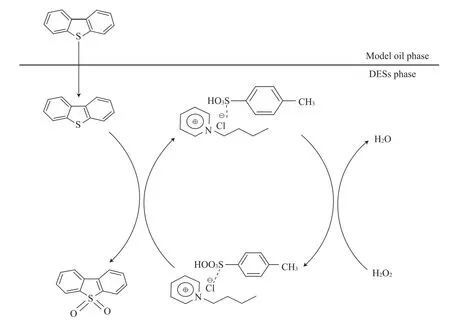
图12 氧化脱硫机理Fig.12 Oxidation desulfurization mechanism.
3 结论
1)通过简单加热[C4Pyr]Cl)和TsOH,制备了一系列[C4Pyr]Cl/nTsOH,(n = 0.1,0.2,0.3)型低共熔溶剂。以[C4Pyr]Cl/nTsOH为催化剂和萃取剂,H2O2为氧化剂组成萃取-催化氧化脱硫体系氧化脱除模拟油中的硫化物。FTIR和1H NMR表征结果显示,[C4Pyr]Cl和TsOH之间形成了氢键。
2)在[C4Pyr]Cl/0.2TsOH加入量1.00 mL、反应温度50 ℃、n(H2O2)∶n(DBT)= 6、模拟油用量5 mL的最佳反应条件下,[C4Pyr]Cl/0.2TsOH对DBT,4,6-DMDBT,BT的脱除率达98.2%,96.0%,40.2%。
3)根据一级动力学方程和Arrhenius方程估算氧化脱除DBT所需的表观活化能约为51.95 kJ/ mol,低于其他文献的报道,说明氧化脱硫反应较易进行。
4)[C4Pyr]Cl/0.2TsOH回收利用5次后,脱硫率略微降低,但仍高达95.1%。[C4Pyr]Cl/0.2TsOH具有高催化活性以及稳定性。
[1] Li Fatang,Liu Ruihong,Wen Jinhua,et al. Desulfurization of dibenzothiophene by chemical oxidation and solvent extraction with Me3NCH2C6H5Cl·2ZnCl2ionic liquid[J].Green Chem,2009,11(6):883-888.
[2] Mei Hai,Mei B W,Yen Teh Fu. A new method for obtaining ultra-low sulfur diesel fuel via ultrasound assisted oxidative desulfurization[J].Fuel,2003,82(4):405-414.
[3] Srivastava V C. An evaluation of desulfurization technologies for sulfur removal from liquid fuels[J].Rsc Adv,2012,2(3):759-783.
[4] Tang Xiaodong,Hu Tao,Li Jinging,et al. Deep desulfurization of condensate gasoline by electrochemical oxidation and solvent extraction[J].Rsc Adv,2015,5(66):53455-53461.
[5] Bösmann A,Datsevich L,Jess A,et al. Deep desulfurization of diesel fuel by extraction with ionic liquids[J].Chem Commun,2001,26(23):2494-2495.
[6] Zhu Wenshuai,Wang Chao,Li Hongping,et al. One-pot extraction combined with metal-free photochemical aerobic oxidative desulfurization in deep eutectic solvent[J].Green Chem,2015,17(4):2464-2472.
[7] Xiong Jun,Zhu Wenshuai,Li Hongping,et al. Few-layered graphene-like boron nitride induced a remarkable adsorption capacity for dibenzothiophene in fuels[J].Green Chem,2015,17(3):1647-1656.
[8] Kulkarni P S,Afonso C A M. Deep desulfurization of diesel fuel using ionic liquids:Current status and future challenges[J].Green Chem,2010,12(7):1139-1149.
[9] Abbott A P,Capper G,Davies D L,et al. Novel solvent properties of choline chloride/urea mixtures[J].Chem Commun,2003,9(1):70-71.
[10] 严楠,熊云奎,夏剑辉,等. 低共熔溶剂中新型螺环吲哚衍生物的绿色合成[J].有机化学,2014,35(2):384-389.
[11] 张盈盈,吉晓燕,陆小华. 胆碱类低共熔溶剂在CO2捕集与分离中的应用[J].化工学报,2014,65(5):1721-1728.
[12] 黄强,左文彬,寸唐湘,等. 基于低共熔溶剂的反溶剂沉淀法制备纳米结构材料[J].实验室研究与探索,2015,34(8):8-10.
[13] Wang Xin,Jiang Wei,Zhu Wenshuai,et al. A simple and cost-effective extractive desulfurization process with novel deep eutectic solvents[J].RSC Adv,2016,6(36):30345-30352.
[14] 李佳慧,胡嘉,赵荣祥,等. 氯化胆碱/草酸型低共熔溶剂氧化脱除模拟油硫化物[J].燃料化学学报,2014,42(7):870-876.
[15] Liu Wei,Jiang Wei,Zhu Wenshuai,et al. Oxidative desulfurization of fuels promoted by choline chloride-based deep eutectic solvents[J].J Mol Catal A:Chem,2016,424:261-268.
[16] Lü Hongying,Li Changping,Deng Chengliang,et al. Deep catalytic oxidative desulfurization(ODS) of dibenzothiophene(DBT) with oxalate-based deep eutectic solvents (DESs)[J]. Chem Commun,2015,51(53):10703-10706.
[17] 方国阳,林金清. 氯化丁基吡啶氯化亚锡离子液体的热稳定分析[C]//第六届全国化学工程与生物化工年会论文集.长沙:全国化学工程与生物化工年会,2010.
[18] 付宏权. 功能化季铵盐离子液体催化地沟油一步法制备生物柴油的研究[D].泉州:华侨大学,2014.
[19] 何志强,鄢浩,王骑虎,等. 温度对氯化胆碱/多元醇型低共熔溶剂物性的影响[J].上海大学学报:自然科学版,2015,21(3):384-392.
[20] Dong Yuxiao,Nie Yi,Zhou Qing. Highly efficient oxidative desulfurization of fuels by Lewis acidic ionic liquids based on iron chloride[J].Chem Eng Technol,2013,36(3):435-442.
[21] Tang Ling,Luo Guangqing,Kang Lihua,et al. A novel[Bmim]PW/HMS catalyst with high catalytic performance for the oxidative desulfurization process[J].Korean J Chem Eng,2013,30(2):314-320.
[22] 张存,马春艳,刘晓勤. 超声强化过氧化氢/三氯乙酸催化氧化柴油深度脱硫研究[J].燃料化学学报,2009,37(3):324-329.
[23] Shi Xianying,Sun Man,Fan Juan,et al. Deep oxidative desulfurization of benzothiophene and dibenzothiophene with a peroxophosphotungstate-ionic liquid brush assembly[J].Appl Organomet Chem,2015,29(9):633-637.
[24] Xie Dong,He Qihui,Su Yangyang,et al. Oxidative desulfurization of dibenzothiophene catalyzed by peroxotungstate on functionalized MCM-41 materials using hydrogen peroxide as oxidant[J].Chin J Catal,2015,36(8):1205-1213.
[25] Otsuki S,Nonaka T,Takashima N,et al. Oxidative desulfurization of light gas oil and vacuum gas oil by oxidation and solvent extraction[J].Energy Fuel,2000,14(6):1232-1239.
[26] Zhu Yunfang,Zhu Mingyuan,Kang Lihua,et al. Phosphotungstic acid supported on mesoporous graphitic carbon nitride as catalyst for oxidative desulfurization of fuel[J].Ind Eng Chem Res,2015,54(7):2040-2047.
[27] Cedeño-Caero L,Gomez-Bernal H,Fraustro-Cuevas A,et al. Oxidative desulfurization of synthetic diesel using supported catalysts:Part Ⅲ. Support effect on vanadium-based catalysts[J].Catal Today,2008,133(1):244-254.
[28] Nie Yi,Dong Yuxiao,Gao Hongshuai,et al. Regulating sulfur removal efficiency of fuels by Lewis acidity of ionic liquids[J].Sci China Chem,2016,59(5):526-531.
[29] Li Liantang,Zhang Jisong,Shen Chun,et al. Oxidative desulfurization of model fuels with pure nano-TiO2as catalyst directly without UV irradiation[J].Fuel,2016,167(1):9-16.
[30] Lü Hongying,Zhang Yongna,Jiang Zongxuan,et al. Aerobic oxidative desulfurization of benzothiophene,dibenzothiophene and 4,6-dimethyldibenzothiophene using an Andersontype catalyst[(C18H37)2N(CH3)2]5[IMo6O24][J].Green Chem,2010,12(11):1954-1958.
[31] Zhao Dishun,Sun Zhimin,Li Fatang,et al. Optimization of oxidative desulfurization of dibenzothiophene using acidic ionic liquid as catalytic solvent[J].J Fuel Chem Technol,2009,37(2):194-198.
[32] Zhou Qin,Fu Shurong,Zou Min,et al. Deep oxidative desulfurization of model oil catalyzed by magnetic MoO3/Fe3O4[J].Rsc Adv,2015,5(85):69388-69393.
[33] 吕志凤,战风涛,李林,等. 用 H2O2-有机酸氧化脱除催化裂化柴油中的硫化物[J].中国石油大学学报:自然科学版,2001,25(3):26-29.
[34] 李晓娜,张薇,徐恩宇,等. Brönsted 酸性离子液体[CMMIm]BF4催化燃油深度脱硫研究[J].东北师范大学学报:自然科学版,2014,46(2):89-92.
(编辑 王 馨)
Preparation of[C4Pyr]Cl/nTsOH deep eutectic solvents and its application in oxidative removal of dibenzothiophene from model oil
Hou Liangpei1,Zhao Rongxiang1,Li Xiuping2
(1. College of Petrochemical Engineering,College of Chemistry,Liaoning Shihua University,Fushun Liaoning 113001,China;2. College of Chemistry & Material Science Engineering,College of Chemistry,Liaoning Shihua University,Fushun Liaoning 113001,China)
A series of acidic deep eutectic solvents[C4Pyr]Cl/nTsOH(n = 0.1,0.2,0.3)were synthesized by the reaction of[C4Pyr]Cl with TsOH via simple heating and stirring. Then[C4Pyr]Cl/nTsOH as extractant and catalyst,H2O2as oxidation were used in extraction-catalytic oxidation desulfurization system. The DESs[C4Pyr]Cl/0.2TsOH and products were characterized by FTIR. Different desulfurization systems,n([C4Pyr]Cl)∶n(TsOH),reaction temperature,n(H2O2)∶n(DBT),[C4Pyr]Cl/0.2TsOH dosage and different type of sulfide on desulfurization efficiency were investigated. According to the experiment results,the optimal reaction conditions were V(DESs) 1.00 mL,50 ℃,n(H2O2)∶n(DBT) 6,V(oil) 5 mL. Under these conditions,the removal for DBT,4,6-DMDBT and BT reached 98.2%,96.0% and 40.2%,respectively. The apparent activation energy for the oxidation desulfurization was about 51.95 kJ/mol which was estimated based on first-order kinetics equation and Arrhenius equation. The DESs could be recycled 5 times,the desulfurization rate is still more than 95.1%.
[C4Pyr]Cl/0.2TsOH;extraction-catalytic oxidation desulfurization;deep eutectic solvents;apparent activation energy
侯良培(1991—),男,河南省商丘市人,硕士生,电邮 18341310461@163.com。联系人:赵荣祥,电话 13470542149,电邮zylhzrx@126.com。
1000-8144(2017)07-0888-08
TQ 426
A
10.3969/j.issn.1000-8144.2017.07.010
2017-01-14;[修改稿日期]2017-04-23。
