Protective e ff ects ofDendrobium nobileLindl. alkaloids on amyloid beta (25–35)-induced neuronal injury
2017-08-07WeiZhangQinWuYanliuLuQihaiGongFengZhangJingshanShi
Wei Zhang, Qin Wu, Yan-liu Lu, Qi-hai Gong, Feng Zhang, Jing-shan Shi
Key Laboratory of Basic Pharmacology of Ministry of Education and Joint International Research Laboratory of Ethnomedicine of Ministry of Education, Zunyi Medical University, Zunyi, Guizhou Province, China
Protective e ff ects ofDendrobium nobileLindl. alkaloids on amyloid beta (25–35)-induced neuronal injury
Wei Zhang, Qin Wu, Yan-liu Lu, Qi-hai Gong, Feng Zhang, Jing-shan Shi*
Key Laboratory of Basic Pharmacology of Ministry of Education and Joint International Research Laboratory of Ethnomedicine of Ministry of Education, Zunyi Medical University, Zunyi, Guizhou Province, China
How to cite this article:Zhang W, Wu Q, Lu YL, Gong QH, Zhang F, Shi JS (2017) Protective e ff ects of Dendrobium nobile Lindl. alkaloids on amyloid beta (25–35)-induced neuronal injury. Neural Regen Res 12(7):1131-1136.
Graphical Abstract
orcid: 0000-0002-1371-6126 (Jing-shan Shi)
Dendrobium nobileLindl. alkaloids (DNLA), the active ingredients of a traditional Chinese medicineDendrobium, have been shown to have anti-oxidative e ff ects, anti-in fl ammatory action, and protective e ff ect on neurons against oxygen-glucose deprivation. However, it is not clear whether DNLA reduces amyloid-beta (Aβ)-induced neuronal injury. In this study, cortical neurons were treated with DNLA at di ff erent concentrations (0.025, 0.25, and 2.5 mg/L) for 24 hours, followed by administration of Aβ25–35(10 μM). Aβ25–35treatments increased cell injury as determined by the leakage of lactate dehydrogenase, which was accompanied by chromatin condensation and mitochondrial tumefaction.e damage caused by Aβ25–35on these cellular properties was markedly attenuated when cells were pretreated with DNLA. Treatment with Aβ25–35down-regulated the expressions of postsynaptic density-95 mRNA and decreased the protein expression of synaptophysin and postsynaptic density-95, all changes were signi fi cantly reduced by pretreatment of cells with DNLA.ese fi ndings suggest that DNLA reduces the cytotoxicity induced by Aβ25–35in rat primary cultured neurons.e protective mechanism that DNLA confers on the synaptic integrity of cultured neurons might be mediated, at least in part, through the upregulation of neurogenesis related proteins synaptophysin and postsynaptic density-95.
nerve regeneration; Dendrobium nobile Lindl. alkaloids; amyloid beta; neurons; synapse; synaptophysin; postsynaptic density-95; cognitive impairment; neuroprotection; neural regeneration
Introduction
Alzheimer’s disease (AD) is the most prevalent mental failure in the elderly. The main clinical manifestation of AD includes a widespread functional disturbance of the central nervous system, characterized by progressive memory impairment, cognitive disorder and altered behavior (Schoemaker et al., 2014). The predominant neuropathological features of AD are senile plaques composed of an aberrant accumulation of amyloid beta (Aβ), neurofibrillary tangles and selective degeneration of neurons and their synapses, generally found in the association cortex and temporal lobe (Zhao et al., 2010; Takeda et al., 2014). It is believed that synaptic injury occurs at a stage prior to the neuronal injury in AD brain (Götz and Ittner, 2008). In addition, the amount ofsynaptic loss and synaptic dysfunction is strongly associated with the degree of cognitive dysfunction (Arendt, 2009).
This suggests that reducing synaptic injury would be an important target for early therapy of AD. Maintaining the morphology and function of synapses depends on the integrity of presynaptic and postsynaptic structures (Luo, 2010). Previous studies have demonstrated that an AD brain contains not only a decrease in density and morphological change of dendritic spines, but also a lower expression of synaptophysin (SYP), postsynaptic density (PSD) and other synapse-associated proteins (Jin and Garner, 2008). Improving the expression of these synapse-associated proteins may preserve the synaptic connections, potentially relieving memory and cognitive impairment. Recently, many investigators have suggested that the overexpression of Aβ, the initiator of a cascade reaction of AD, can induce AD-related synaptic dysfunction at an early stage of the disease. This disturbs neurogenesis, and eventually leads to cognitive problem (Senthilkumar et al., 2013; Welzel et al., 2014).
Dendrobium nobileLindl. alkaloids (DNLA) are alkaloids from a traditional Chinese herbal medicineDendrobium, which is used to treat cataracts and throat inflammation (Zhang et al., 2003). The major active pharmacological ingredients have been reported to be alkaloids, stilbenoids, glycosides and polysaccharides (Pan et al., 2013).Dendrobiumextracts possess a broad spectrum of pharmacological activities, including anti-aging, anti-free radical injury, anti-inflammatory and immunomodulatory effects, and anti-tumor function (Wei, 2005). Our previous research had demonstrated that DNLA has neuroprotective e ff ects. DNLA can inhibit lipopolysaccharide-induced astrocyte activation and reduce the production of pro-in fl ammatory factors such as interleukin-6 and tumor necrosis factor-α (Zhang et al., 2011). In addition, DNLA can significantly protect against oxygen-glucose deprivation/reperfusion (OGD/RP) induced neuronal damage in rat primary neuron cultures (Wang et al., 2010). In this study, we used Aβ25–35to disrupt synaptic function, mimicking the early stages of AD, to further investigate the neuroprotective function of DNLA on a model of Aβ-induced neurotoxicity in rat primary neuron cultures. We measured the expression of SYP and postsynaptic density-95 (PSD-95), which may be involved in the mechanism of DNLA-elicited neuroprotective function.
Materials and Methods
Animals
One-day-old Sprague-Dawley rats (speci fi c-pathogen-free II, 30 females and 10 males, aged 6–7 weeks, weighing 180–200 g) were purchased from the Animal Center of the Third Military Medical University of China (certi fi cate No. SCXK (Jun) 2007-0005).e rats were maintained in an air-conditioned animal facility at 23 ± 1°C and a 12-hour light/dark cycle. Rats were given free access to water and food.e experiment was carried out in strict accordance with the State Committee of Science and Technology of the People’s Republic of China Order on November 14, 1988 (revised 2011), and the study protocol followed the Regulations of the 27thAugust 2007 approved by the Animal Experimental Ethics Committee of the Zunyi Medical University of China.
Cell culture and identi fi cation
Primary neuronal cultures were prepared from the cortex of 1-day-old Sprague-Dawley rat pups by enzymatic digestion (Li et al., 2013). Cells dissociated from the cerebral cortices were collected in Dulbecco’s modified eagle medium/nutrient mixture F12 (DMEM/F12; Gibco BRL, Grand Island, NY, USA) containing 10% fetal bovine serum (Sigma Chemical Co., St. Louis, MO, USA), 10% horse serum (Gibco BRL), and 100 U/mL penicillin/streptomycin.e cells were plated at a density of 1 × 105/mL, seeded in 24- and 6-well culture plates pre-coated with poly-D-lysine (Sigma Chemical Co.). Cultures were maintained at 37°C in a humidi fi ed incubator of 5% CO2and 95% air. Aer seeding for 24 hours, cultures were replaced with maintenance medium containing Neurabasal-A (Gibco BRL) supplemented with 2% B27 (Gibco BRL). The cortical neurons were immunostained with an antibody against neuron-speci fi c enolase (NSE) (Wuhan Boster Biological Technology, Wuhan, China) and veri fi ed by morphologic examination.
Drug intervention
The 7-day-old neuronal cell cultures were used for drug treatments. Cells were homogeneously distributed between each treatment group. Five groups were set up for cell culture experiments, which were: I Control group (untreated cells: no Aβ25–35or DNLA), II Aβ25–35treated group (10 μM Aβ25–35(Sigma Chemical Co.) for 24 hours), DNLA pretreated groups (treatment with DNLA at concentrations of 0.025, 0.25, or 2.5 mg/L, III – V respectively, for 24 hours, followed by 10 μM Aβ25–35induction for 24 hours). DNLA (purity ≥86.3%) was provided by the pharmacology laboratory of Zunyi Medical College (Guizhou, China).
Morphological examination
Leakage of lactate dehydrogenase (LDH)
Real-time reserve transcription-polymerase chain reaction (RT-PCR)
Immunocytochemical staining
Cortical neuronal cells were fixed with 4% paraformaldehyde for 1 hour. Afterwards cells were incubated with a rabbit monoclonal anti-SYP antibody (1:125; cat. on. ab32127, Abcam, Cambridge, UK) for 1 hour at 37°C. The primary antibodies were diluted in 0.1 M phosphate buffered saline (PBS) containing 0.5% bovine serum albumin and 0.5% Triton X-100.e secondary antibodies used were Texas Red donkey anti-rabbit IgG antibody (cat. on. P0179, 1:1,000; Beyotime Institute of Biotechnology, Nanjing, China) for 30 minutes at 37 °C.e fl uorescence was observed using a light microscope with the appropriate fl uorescence fi lters.e integrated optical density of active SYP-positive expression was determined using the Imagepro-Plus 6.0 soware (Media Cybernetics, Inc., Rockville, MD, USA) in fi ve fi elds in each group.
Western blot assay
Cortical neuronal cells were washed with cold PBS, and lysed with radioimmune precipitation assay lysis bu ff er consisting of 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1 mM ethylenediamine tetraacetic acid, 1% Triton X-100, 1% sodium deoxycholate, 1 mM Na3VO4, 1 mM phenylmethylsulfonyl fl uoride, and a freshly-added protease inhibitor cocktail.e cytosols were prepared by centrifugation at 12,000 ×gfor 30 minutes at 4°C. Protein concentrations were quanti fi ed with the bicinchoninic acid assay (Sigma-Aldrich, St. Louis, MO, USA) (Leinenga and Götz, 2015). Equal amounts of total protein (15–30 μg) were subjected to electrophoresis on 5–12% Bis-Tris Nu-PAGE gels (Invitrogen), followed by electrophoretic transfer onto polyvinylidene di fl uoride membranes. Membranes were blocked with 5% nonfat milk for 1 hour, then incubated with the primary antibodies, mouse monoclonal anti-PSD95 (1:100; Abcam), rabbit polyclonal anti-β-actin (1:1,000; Beyotime Institute of Biotechnology), or rabbit monoclonal anti-SYP (1:10,000; Abcam) overnight at 4°C. After warming, membranes were incubated with horseradish peroxidase-conjugated secondary antibodies, goat anti-mouse IgG and goat anti-rabbit IgG (1:1,000; Beyotime Institute of Biotechnology) for 1 hour at room temperature.e immunoblots were visualized with enhanced chemiluminescence substrate. Images were captured using a ChemiXRS system (Bio-Rad Laboratories, Inc., Richmond, CA, USA).e bands were scanned and analyzed using optical density values with background subtraction and normalized to β-actin using Quantity One soware v4.52 (Bio-Rad Laboratories, Inc., Richmond, CA, USA).
Statistical analysis
All data are presented as the mean ± SD. Statistical significance was determined using analysis of variance with the Prism 5 soware (GraphPad; GraphPad Soware, Inc., CA, USA) followed by Dunnett’st-test.P< 0.05 was considered statistically signi fi cant.
Results
Morphological change of Aβ25–35-induced cortical neurons aer DNLA intervention
All the cortical neurons were prepared from 1-day-old rat pups using enzymatic digestion.e neurons were veri fi ed by morphology and staining with anti-NSE antibody (Figure 1). Populations of neurons typically with purity greater than 85% were used in the experiments. The cortical neurons were cultured continuously for 7 days. As shown in Figure 1, these cells displayed good refractive and stereoscopic properties of neuron cell body, as well as complete synaptic structures.e 7-day-old neurons were then used for drug treatments to study the protective e ff ects of DNLA.
Ultrastructural examination of cortical neurons showed that in the control group, cell membrane and cell organelles were complete, and chromatin was distributed homogeneously (Figure 2A). Aer Aβ25–35treatment, chromatin was condensed and clumped, and mitochondria were swollen and vacuolar as shown in Figure 2B. Pre-treatment with DNLA signi fi cantly reduced the levels of neuronal damage compared with those of the Aβ25–35treated group. As shown in Figure 2C–E, the neurons pretreated with DNLA displayed clearer membrane structures of mitochondria and rough endoplasmic reticulum than the cells from Aβ25–35treated group (Figure 2C–E).
DNLA e ff ects on leakage of LDH of Aβ25–35-induced cortical neurons
Exposure of cultured neurons to Aβ25–35produced a significant increase in neuronal injury compared to the control group, as evidenced by an increase in LDH leakage (P< 0.01). DNLA treatment markedly attenuated LDH leakage (P<0.05), indicating a protective role of DNLA against Aβ25–35-induced neuronal damage (Figure 3).
DNLA e ff ects on expression of PSD-95 mRNA in Aβ25–35-induced cortical neurons
To understand the molecular events linking to the DNLAmediated neuroprotective mechanism, we investigated the expression level of PSD-95, a major scaffolding protein in ensuring the integrity of the neuronal synapse and receptors participating in information flow (Chen et al., 2015). As shown in Figure 4, Aβ25–35signi fi cantly reduced the expression of PSD-95 mRNA by approximately 50% compared with the control group. However, in the DNLA (0.25 mg/L) pretreated neurons, the expression levels of PSD-95 mRNA were significantly higher compared to the expression of PSD-95 aer Aβ25–35treatment alone (P< 0.05) (Figure 4).
DNLA e ff ects on the expression of SYP and PSD-95 protein in Aβ25–35-induced cortex neuron cells
As shown in Figure 5A–C, Aβ25–35challenge resulted in severe synaptic damage, and loss of synaptic connections. Immunoreactivity of SYP-positive expression decreased significantly compared with that in the control group (P< 0.01). These morphological changes were significantly attenuated by the pretreatment with DNLA in a dose-dependent manner. The immunoreactivity of positive expression increased signi fi cantly in DNLA groups compared with Aβ25–35treated group (DNLA 0.25 mg/L group,P< 0.05; DNLA 2.5 mg/L group,P< 0.01). We further determined the protein expression of SYP and PSD-95 by western blot assay. As shown in Figure 5C, Aβ25–35treatment decreased signi fi cantly the expression of SYP (P<0.05) and PSD-95 (P< 0.01) proteins compared with the control group, while the expression levels of both proteins were enhanced with pretreatment of DNLA as compared to Aβ25–35treatment alone (P< 0.05), suggesting a role of SYP and PSD-95 in DNLA-mediated neuroprotective mechanism.
Discussion
In this study, we clearly demonstrated that DNLA has a neuroprotection role against Aβ25–35-induced cytotoxicity in primary cultured rat cerebral cortical neurons. DNLA significantly attenuated the damage to morphological structures and inhibited the release of LDH caused by Aβ25–35in cortical neurons. Furthermore, we showed that pretreatment of DNLA signi fi -cantly blocked the decrease in the expression of both synaptic proteins SYP and PSD-95. These molecular changes may be central to the DNLA-mediated synaptic protection.
A wealth of evidence indicates that accumulation of excessive intraneuronal Aβ initiates a complex cascade of neurotoxic alterations (Gaspar et al., 2010). Aβ, a main component of diffuse plaques in AD brain, results in the production of neurotoxins and causes synaptotoxic e ff ects, leading ultimately to degeneration of neurites and neuronal cell bodies. These pathological changes result in cognitive impairment and the development of dementia (Pike et al., 2011). Aβ is not only the origin but also the common pathway in the occurrence and development of AD. Inhibition of these Aβ-induced toxic e ff ects has been a key area in the study of the pathological progression and in the exploration of effective treatments for AD. In the present study, we observed that Aβ25–35elicited neuronal toxicity, including an increase in the leakage of LDH and disruption of neuronal cell organelles. However, these cytotoxic effects were significantly prevented when cells were pretreated with DNLA. Clearly, our data demonstrated that DNLA possesses a neuroprotective e ff ect on cortical neurons.
Growing attention in the recent decade has been paid to synaptic dysfunction due to the synaptotoxic e ff ects of Aβ. Neuropathological studies in humans have supported the findings that synaptic loss occurs at an earlier stage of AD than gross neuronal injury (Götz and Ittner, 2008; Marcello et al., 2012). In addition, there was a positive correlation between the degree of synaptic loss and synaptic dysfunction and the cognitive impairment in AD patients (Clare et al., 2010). All these suggest the important roles of synaptic structure and function in the development of AD.
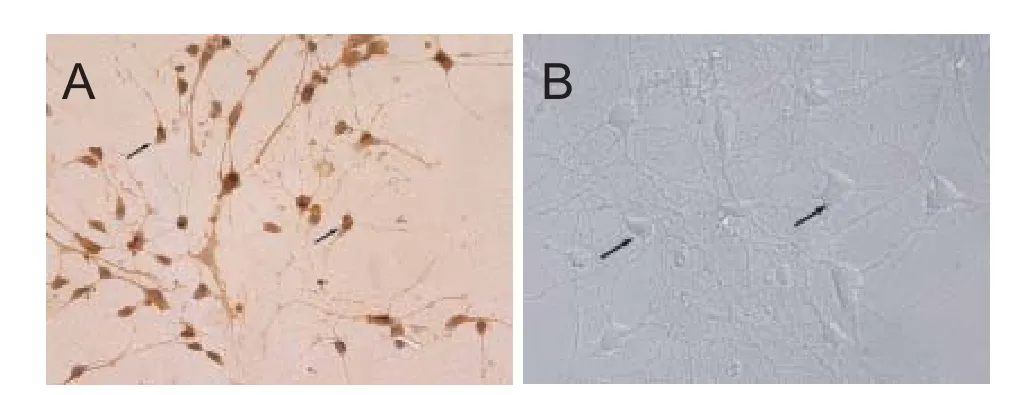
Figure 1 Identi fi cation of cortical neurons (× 400).
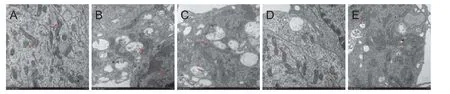
Figure 2 E ff ects of DNLA administration on the ultrastructural changes induced by Aβ25–35in rat primary cultured neurons detected by transmission electron microscope.
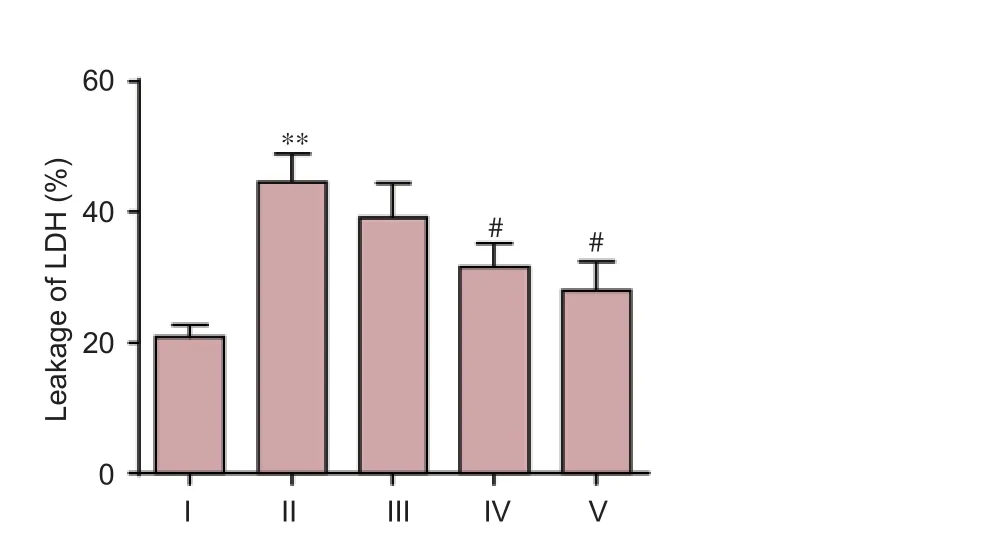
Figure 3 DNLA e ff ects on Aβ25–35-induced leakage of LDH in rat primary cultured neurons.Rat primary cultured neurons were pretreated with DNLA for 24 hours before the addition of Aβ25–35. 24 hours later, the leakage of LDH was determined by biochemical instrument. Results are expressed as the mean ± SD from three-independent experiments, and analyzed by analysis of variance followed by Dunnett’st-test. **P< 0.01,vs. control group; #P< 0.05,vs. Aβ25–35treated group. I: Control group, without treatment; II: Aβ25–35group, treated with 10 μM Aβ25–35for 24 hours; III–V: Neuronal cultures pretreated with 0.025, 0.25, 2.5 mg/L DNLA, respectively, for 24 hours, followed by 10 μM Aβ25–35administration for 24 hours. DNLA:Dendrobium nobileLindl. alkaloids; Aβ25–35: amyloid-beta (25–35); LDH: lactate dehydrogenase.
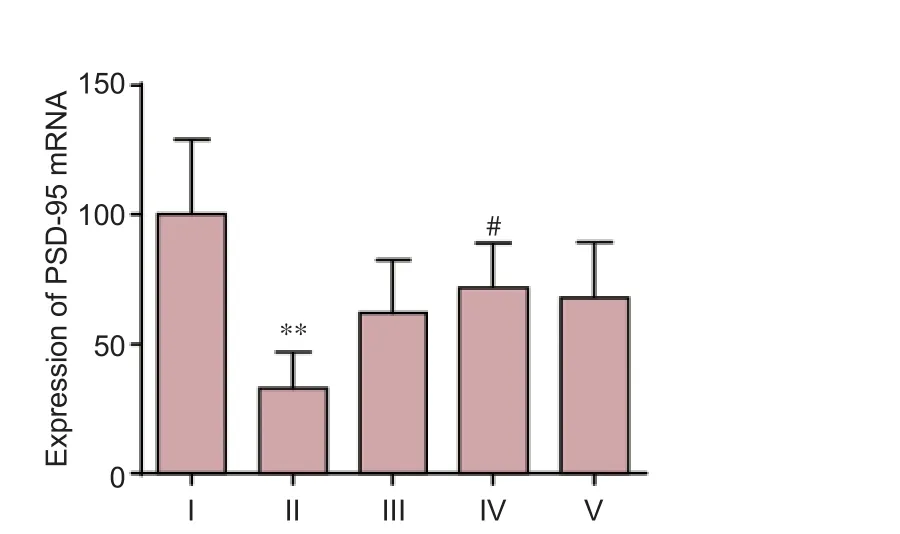
Figure 4 E ff ects of DNLA and Aβ25–35on mRNA expression of PSD-95 in rat primary cultured neurons.Rat primary cultured neurons were pretreated with DNLA for 24 hours and then treated with Aβ25–35.e mRNA expression of PSD-95 was measured by real-time reverse transcription-polymerase chain reaction. Results are expressed as the mean ± SD from fi ve independent experiments, and analyzed by analysis of variance followed by Dunnett’st-test. **P< 0.01,vs. control group; #P< 0.05,vs. Aβ25–35-treated group. I: Control group, without treatment; II: Aβ25–35-treated group, treated with 10 μM Aβ25–35for 24 hours; III–V: Neuron cultures treated with 0.025, 0.25, 2.5 mg/L DNLA, respectively, for 24 hours, followed by 10 μM Aβ25–35intoxication for 24 hours. DNLA:Dendrobium nobileLindl. alkaloids; Aβ25–35: amyloid-beta (25–35); PSD-95: postsynaptic density-95.
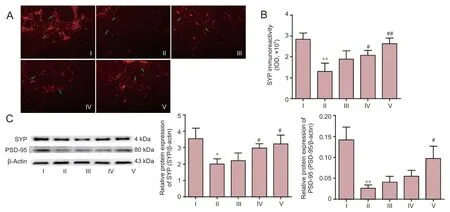
Figure 5 E ff ects of DNLA on Aβ25–35–induced protein expression of synapse-associated proteins in rat primary cultured neurons.
SYP is one of the major glycosylated protein found in presynaptic vesicles. It has been considered as a speci fi c marker for the nerve terminals (Canas et al., 2014). In addition, SYP is a marker for the functionality of the synapse. It is also involved in the process of exocytotic release of neurotransmitter and membrane recycling in nerve terminals (Vukic et al., 2009; Gordon et al., 2011). In this study, we examined both the density and distribution of SYP in order to reflect the synaptic density and distribution of the synapse.
PSD-95, a postsynaptic sca ff olding protein, is distributed evenly throughout the PSD which is a complex, disk-shaped structure attached to the postsynaptic membrane at synapses (Chen et al., 2011). PSD-95 binds to the glutamate receptors and to other components of the PSD, including Ca2+/calmodulin-dependent protein kinase II (CaMKII), Shank, and actin (Chen et al., 2011). Several studies have indicated the involvement of PSD-95 in the tra ffi cking of α-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid receptors, and gating of N-methyl-D-aspartate receptors (Opazo et al., 2012; Genevieve et al., 2014). Tra ffi cking of receptors at the PSD results in changes in synaptic transmission and structure, and these changes are responsible for the formation, function, and plasticity of spine synapses.
In this study, the results of immunocytochemistry andwestern blot assay suggest that DNLA e ff ectively suppressed the decrease in the level of density and expression of SYP protein caused by Aβ25–35treatment. After treatment with DNLA, the levels of PSD-95 mRNA and protein in primary cultured neurons were enhanced.is suggests that DNLA may play a role in maintaining the integrity of pre-synaptic and post-synaptic structures, thereby improving the synaptic connection, synaptic transmission and synaptic plasticity.ese molecular events associated with DNLA might play a key role in improving the information transmission of the nervous system that is involved in the mechanism of DNLA-mediated neuroprotective mechanism.
In summary, DNLA protected cortical neurons against Aβ25–35-mediated neurotoxicity and synaptic damage. This synaptic protective effect was at least partially mediated by the reduced Aβ25–35-induced decrease in SYP and PSD-95. Thus, DNLA might possess a potential benefit for the prevention and early therapy of AD. Further studies are required to fully de fi ne the underlying molecular mechanism of DNLA elicited neuroprotection. This work provides a pharmacological basis for DNLA in prevention and treatment of AD, especially in the early stage of AD.
Author contributions:WZ and QW performed experiments. WZ wrote most of the text of the article, edited the figure and figure legend, and provided references. YLL contributed to the collection and analysis of the experimental data. QHG and FZ provided critical commentary and assistance in technique application. JSS designed the study, reviewed the document and was responsible for fundraising. All authors approved the fi nal version of the paper.
Con fl icts of interest:None declared.
Open access statement:
Contributor agreement:A statement of “Publishing Agreement” has been signed by an authorized author on behalf of all authors prior to publication.
Plagiarism check:This paper has been checked twice with duplication-checking soware ienticate.
Peer review:A double-blind and stringent peer review process has been performed to ensure the integrity, quality and signi fi cance of this paper.
Arendt T (2009) Synaptic degeneration in Alzheimer’s disease. Acta Neuropathol 118:167-179.
Canas PM, Simões AP, Rodrigues RJ, Cunha RA (2014) Predominant loss of glutamatergic terminal markers in a β-amyloid peptide model of Alzheimer’s disease. Neuropharmacology 76 Pt A:51-56.
Chen X, Levy JM, Hou A, Winters C, Azzam R, Sousa AA, Leapman RD, Nicoll RA, Reese TS (2015) PSD-95 family MAGUKs are essential for anchoring AMPA and NMDA receptor complexes at the postsynaptic density. Proc Natl Acad Sci U S A 112:E6983-E6992.
Chen X, Nelson CD, Li X, Winters CA, Azzam R, Sousa AA, Leapman RD, Gainer H, Sheng M, Reese TS (2011) PSD-95 is required to sustain the molecular organization of the postsynaptic density. J Neurosci 31:6329-6338.
Clare R, King VG, Wirenfeldt M, Vinters HV (2010) Synapse loss in dementias. J Neurosci Res 88:2083-2090.
Götz J, Ittner LM (2008) Animal models of Alzheimer’s disease and frontotemporal dementia. Nat Rev Neurosci 9:532-544.
Gaspar RC, Villarreal SA, Bowles N, Hepler RW, Joyce JG, Shughrue PJ (2010) Oligomers of β-amyloid are sequestered into and seed new plaques in the brains of an AD mouse model. Exp Neurol 223:394-400.
Genevieve L, Andre V, Rudolf K, Eric T, Beat Michel R, Armand S (2014) Pathological reorganization of NMDA receptors subunits and postsynaptic protein PSD-95 distribution in Alzheimer’s disease. Curr Alzheimer Res 11:86-96.
Gordon SL, Leube RE, Cousin MA (2011) Synaptophysin is required for synaptobrevin retrieval during synaptic vesicle endocytosis. J Neurosci 31:14032-14036.
Jin Y, Garner CC (2008) Molecular mechanisms of presynaptic di ff erentiation. Annu Rev Cell Dev Biol 24:237-262.
Koh JY, Choi DW (1987) Quantitative determination of glutamate mediated cortical neuronal injury in cell culture by lactate dehydrogenase effl ux assay. J Neurosci Methods 20:83-90.
Leinenga G, Götz J (2015) Scanning ultrasound removes amyloid-β and restores memory in an Alzheimer’s disease mouse model. Sci Transl Med 7:278ra233.
Li D, Qiu Z, Shao Y, Chen Y, Guan Y, Liu M, Li Y, Gao N, Wang L, Lu X, Zhao Y, Liu M (2013) Heritable gene targeting in the mouse and rat using a CRISPR-Cas system. Nat Biotech 31:681-683.
Luo ZG (2010) Synapse formation and remodeling. Sci China Life Sci 53:315-321.
Marcello E, Epis R, Saraceno C, Di Luca M (2012) ynaptic dysfunction in Alzheimer’s disease. Adv Exp Med Biol 970:573-601.
Opazo P, Sainlos M, Choquet D (2012) Regulation of AMPA receptor surface di ff usion by PSD-95 slots. Curr Opin Neurobiol 22:453-460.
Pan LH, Feng BJ, Wang JH, Zha XQ, Luo JP (2013) Structural characterization and anti-glycation activity in vitro of a water-soluble polysaccharide from dendrobium huoshanense. J Food Biochem 37:313-321.
Pike KE, Ellis KA, Villemagne VL, Good N, Chételat G, Ames D, Szoeke C, Laws SM, Verdile G, Martins RN, Masters CL, Rowe CC (2011) Cognition and beta-amyloid in preclinical Alzheimer’s disease: Data from the AIBL study. Neuropsychologia 49:2384-2390.
Schoemaker D, Gauthier S, Pruessner JC (2014) Recollection and familiarity in aging individuals with mild cognitive impairment and Alzheimer’s disease: a literature review. Neuropsychol Rev 24:313-331.
Senthilkumar S, Aaron T, Jayakumar R (2013) Pathogenesis of Abeta oligomers in synaptic failure. Curr Alzheimer Res 10:316-323.
Takeda S, Sato N, Morishita R (2014) Systemic in fl ammation, bloodbrain barrier vulnerability and cognitive/non-cognitive symptoms in Alzheimer disease: relevance to pathogenesis and therapy. Front Aging Neurosci 6:171.
Vukic V, Callaghan D, Walker D, Lue LF, Liu QY, Couraud PO, Romero IA, Weksler B, Stanimirovic DB, Zhang W (2009) Expression of infl ammatory genes induced by beta-amyloid peptides in human brain endothelial cells and in Alzheimer’s brain is mediated by the JNKAP1 signaling pathway. Neurobiol Dis 34:95-106.
Wang Q, Gong Q, Wu Q, Shi J (2010) Neuroprotective e ff ects of Dendrobium alkaloids on rat cortical neurons injured by oxygen-glucose deprivation and reperfusion. Phytomedicine 17:108-115.
Wei XY (2005) Review on the research progress of alkaloid in dendrobium plants. Zhongguo Yaoshi 19:445-447.
Welzel AT, Maggio JE, Shankar GM, Walker DE, Ostaszewski BL, Li S, Klyubin I, Rowan MJ, Seubert P, Walsh DM, Selkoe DJ (2014) Secreted amyloid β-proteins in a cell culture model include N-terminally extended peptides that impair synaptic plasticity. Biochemistry 53:3908-3921. Zhang GN, Bi ZM, Wang ZT, Xu LS, Xu GJ (2003) Advances in studies on chemical constitutents from plants of Dendrobium Sw. Zhongcaoyao 34:U005-U008.
Zhang JQ, Wu Q, Gong QH, Zhang F, Jin F, Nie J, Shi JS (2011) E ff ects of Dendrobium nobile total alkaloids on lipopolysaccharide-induced astrocyte activation and pro-inflammatory factors production. Zhongguo Yaolixue Tongbao 27:824-827.
Zhao WQ, Santini F, Breese R, Ross D, Zhang XD, Stone DJ, Ferrer M, Townsend M, Wolfe AL, Seager MA, Kinney GG, Shughrue PJ, Ray WJ (2010) Inhibition of calcineurin-mediated endocytosis and alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors prevents amyloid beta oligomer-induced synaptic disruption. J Biol Chem 285:7619-7632.
Copyedited by Yu J, Li CH, Qiu Y, Song LP, Zhao M
10.4103/1673-5374.211193
Accepted: 2017-04-24
*Correspondence to: Jing-shan Shi, Ph.D., shijs@zmc.edu.cn.
杂志排行

中国神经再生研究(英文版)的其它文章
- SoxC transcription factors in retinal development and regeneration
- Umbilical cord: an unlimited source of cells di ff erentiable towards dopaminergic neurons
- Targeting 14-3-3 adaptor protein-protein interactions to stimulate central nervous system repair
- RACK1 regulates neural development
- Schwann cell development, maturation and regeneration: a focus on classic and emerging intracellular signaling pathways
- BDNF pro-peptide: a novel synaptic modulator generated as an N-terminal fragment from the BDNF precursor by proteolytic processing