RIP1/3对创伤性脑损伤后神经元的作用及其机制研究
2017-08-07王宏斌郑新瑞袁华何芳梅
王宏斌 郑新瑞 袁华 何芳梅
RIP1/3对创伤性脑损伤后神经元的作用及其机制研究
王宏斌 郑新瑞 袁华 何芳梅
目的 探讨RIP1/3对创伤性脑损伤(TBI)后神经元的影响及其作用机制。方法 将体外培养的皮层神经元分为4组:siRIP组、Nec-1预处理组、阴性质粒转染组和对照组。通过慢病毒转染法分别干涉RIP1、RIP3表达,建立离体TBI损伤模型,通过流式细胞学检测划伤后神经元存活情况。对体外培养的皮层神经元敲除RIP1/3,建立谷氨酸诱导神经元损伤模型,通过流式细胞学检测谷氨酸刺激后神经元存活情况。结果 siRIP组及Nec-1预处理组机械性损伤后神经元存活率高于阴性质粒转染组和对照组。Nec-1预处理组谷氨酸损伤后神经元存活率高于对照组,而siRIP组存活率与对照组和阴性转染组比较未见显著差异。结论 RIP1和RIP3可能对TBI诱导后神经元死亡有作用,而RIP1抑制剂Nec-1可能对TBI具有脑保护作用。RIP1/3与谷氨酸兴奋毒性诱导细胞死亡无关,而Nec-1对于谷氨酸损伤具有潜在保护作用,并可能存在特异性靶点。
创伤性脑损伤 受体相互作用蛋白1/3 神经元 谷氨酸
创伤性脑损伤(Traumatic brain injury,TBI)是常见的外科损伤,也是神经外科的重要临床疾病之一。全世界每年有近1亿人因TBI而死亡或者住院,在目前现存人口中大约有60亿人次曾经历过TBI[1-3],这给社会及民众都造成了巨大的经济和精神负担。颅脑外伤中脑组织会先后经历初次脑损伤和二次脑损伤。初次脑损伤往往时间迅速、难以防治,但相对的危害也较小,而继发性脑损伤其中包括初次创伤后机体所发生的一系列病理生理反应如细胞凋亡、坏死、兴奋性神经递质毒性释放、炎症反应、氧自由基生成、神经脱髓鞘、白质病变以及癫痫等,因其机制复杂,并且易产生级联效应,因而危害更大,可导致长期的运动和认知功能障碍。
受体相互作用蛋白(RIP)家族中的RIP1和RIP3可以诱导多种不同的细胞转归。已有研究证实其在细胞坏死与凋亡中发挥着重要作用,在炎症因子等因素的刺激下RIP激活自身丝苏氨酸激酶活性,从而介导细胞死亡。已有研究证实RIP蛋白在神经系统缺血和退行性变中有作用,但关于其对TBI的影响,因为机制复杂,而鲜有研究。因此,本研究的目的旨在初步探索TBI后RIP1及RIP3的表达变化以及RIP分子对于TBI后神经元的影响及作用。
1 材料与方法
1.1 材料及主要仪器
试验孕2周昆明小鼠(25~30 g)培养于第四军医大学动物中心;FBS、Neurobasal培养液、10%胎牛血清、B27添加剂、DMEM购自美国Gibco公司;PBS购自西安化学试剂厂;PLL、L-谷氨酰胺、多聚赖氨酸、胰蛋白酶购自美国Sigma公司;NF200多克隆抗体购自武汉博士得公司;LDH检测试剂盒购自美国 BioVision 公司;5% CO2孵箱购自美国Forma公司;高速低温离心机购自美国Beckman公司;光学解剖显微镜购自德国Leica公司;恒温孵箱购自上海市仪器厂;电热恒温水浴锅购自上海市医疗仪器厂;纯水机购自美国Millipore公司;精密电子天平购自瑞士Mteeler公司;医用离心机购自上海医疗仪器厂;透射电镜购自日本Olympus公司;倒置相差显微镜购自德国Leica公司;UV2300分光光度计购自上海天美科学仪器厂。
1.2 方法
1.2.1 细胞培养
于光学解剖显微镜下逐一剥离胎鼠的皮层结构。在4 ℃预冷的新鲜PBS液中剥去软、硬脑膜和周围血管组织;剥离后用D-Hanks液漂洗皮层,时间为5 min;用眼科剪剪碎漂洗过的组织,加入胰蛋白酶消化液(1.25 g/L,2 mL),置于CO2浓度为5%的38 ℃恒温孵箱中进行细胞消化,时间为20 min;室温条件下用移液器吸去上层剩余的消化液,将消化后的脑组织转移至含有DMEM与20%胎牛血清的无菌离心管中,静置10 min,静置后终止消化,用DMEM培养液再次漂洗细胞,时间5 min;使用胶头滴管室温下在DMEM细胞培养液(含20%胎牛血清)中将消化后的脑组织碎片缓慢吹打成为细胞悬液,静置5 min后显微镜下观察滴于细胞悬液计数板并计数细胞;预先用多聚赖氨酸包被60 mm培养皿1 d,之后用PBS液反复漂洗3~4遍,室温下晾干备用,以4×105/cm2将细胞接种于35 mm培养皿上,培养液中加入终浓度为100 000 U/L的青-链霉素和谷氨酰胺(0.6 mmo1/L);将细胞置于恒温孵箱(设定同上)中进行培养,时间24 h,次日弃去青-链霉素培养液,用的DMEM培养液38 ℃下漂洗细胞2次,并更换为等量的含B27(20 m1/L)的Neurobasal培养液;每日于显微镜下观察神经元生长状况并拍照记录;每隔3 d半量更换Neurobasal培养液。
1.2.2 建立离体TBI损伤模型
神经元体外培养第6 d将培养液替换成新鲜的不含抗生素的完全培养液;24 h后对于35 mm培养皿培养的神经元,将1×109TU/ml siRIP1或1×109TU/ml siRIP3分别加入到10 μL病毒稀释液中,轻轻混匀,然后在细胞培养液中缓慢加入转染试剂复合物并均匀混合,随后38 ℃下50 ml/L CO2孵箱中孵育72 h以上。对照组仅替换培养液继续培养。神经元培养在第11 d将对照组和基因转染组再各分2组,一半提前1 d加入使用浓度为50 μM的necrostatin-1,各组进行相应的试验干预处理,每组6个培养皿;神经元在转染空载体或siRIP1/3后72 h进行损伤;损伤方法采用Fаden等的实验方法[4],用10 μL微量移液器塑料枪头,快速地机械性划割,划伤速度与力度尽量保持一致,以保证各组神经元损伤程度和范围大体相同;采用本法造成的损伤模型具有较高的稳定性,能够近似地在神经元上模拟机械性脑损伤的各种病理生理过程[5]。由此离体神经元TBI损伤模型分为4个小组,分别是siRIP组(分为RIP1干扰质粒转染组及RIP3干扰质粒转染组)、Nec-1预处理组、阴性质粒转染组和单纯损伤组。
1.2.3 转染siRIP1/3后对神经元TBI损伤后细胞死亡率研究分析
神经元转染siRIP1与siRIP并进行损伤24 h后采用Cell titer glo试剂盒检测,此试剂盒是利用对ATP水平的检测来表明细胞存活率,具体方法如下:于96孔培养板中需要检测的孔内加入20 uL的Cell titer glo 试剂,摇床上震荡5 min,静置10 min后用多功能酶标仪检测化学发光。
1.2.4 流式细胞术检测机械损伤神经元凋亡和坏死
用6孔板培养细胞,神经元培养成功后根据各组要求建模,继续培养,待细胞生长达到50%~70%;将培养液移至离心管内,用PBS洗涤贴壁细胞1次,并加入适量胰酶消化细胞;室温孵育,可使贴壁细胞轻轻吹打下来时吸除细胞消化液;务必避免胰酶过度消化细胞;贴壁细胞2 000 r/min消化离心5 min并收集;用PBS洗涤细胞2次2 000 r/min离心5 min收集1~5×105细胞;加入100 μL的1×Binding Buffer以悬浮细胞;加入5 μL Annexin V-PE和10 μL 7-AAD染液混匀,室温、避光、反应15 min;补加380 μL的1×Binding Buffer,混匀;在1 h内进行流式细胞仪的观察和检测。
1.2.5 体外模型中干涉RIP1/3对谷氨酸诱导神经元死亡模型的影响
将体外培养的皮层神经元敲除RIP1/3。建立谷氨酸诱导神经元损伤模型。神经元培养的第11 d将对照组和基因转染组再各分2组,各组进行相应的试验干预处理,每组6个培养皿;神经元在转染空载体或siRIP1/3后72 h进行谷氨酸损伤:加入谷氨酸500 μmol/L与10 μmol/L甘氨酸预处理12 h的混合液刺激神经元15 min后更换为正常培养液,过夜培养;转染siRIP1/3后对神经元谷氨酸损伤后细胞死亡率进行分析。
1.2.6 统计学处理

2 结 果
2.1 RIP1/3干扰质粒转染神经元
通过使用干扰RIP1与RIP3慢病毒载体转染神经元发现,绿色荧光显示病毒干扰效率较高,WB显示过慢病毒干扰后RIP1与RIP3表达明显下调(图1)。
2.2 神经元存活情况
TBI后阴性质粒转染组存活细胞明显减少(36±3.8)%,而干扰RIP1或RIP3都可使存活率增加(72%,68%)。同时,使用Nec-1也有明显效果(P<0.05)(图2)。

注:1为单纯损伤组;2为阴性质粒转染组;3为RIP1干扰质粒转染组;4为RIP3干扰质粒转染组;5为Nec-1处理组图1 RIP1/3干扰质粒转染神经元分析
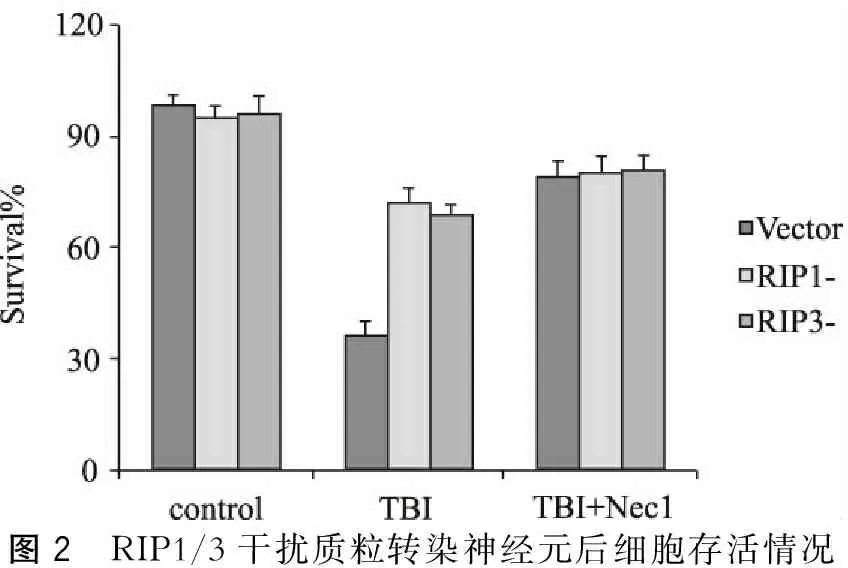
图2 RIP1/3干扰质粒转染神经元后细胞存活情况
2.3 流式细胞术检测
流式细胞术检测显示阴性质粒转染组与单纯损伤组无显著差异,干扰RIP1与RIP3后细胞凋亡与坏死与损伤组相比显著减少(P<0.01),而使用Nec-1组细胞存活率较干扰组更高(P<0.05)(图3)。
2.4 谷氨酸诱导神经元死亡模型的细胞生存率
在对体外神经元以谷氨酸刺激后神经元生存率明显下降(17.8±2)%,同时可以看到干扰RIP1或干扰RIP3对于神经元谷氨酸损伤细胞死亡率都无明显影响,而同时添加Nec-1预处理组死亡率明显下降(P<0.01)。使用炎症因子TNF-α诱导离体神经元凋亡模型中干扰RIP1与干扰RIP3组生存率都明显上升(P<0.01),与Nec-1保护效果无显著差异(图4~5)。
3 讨 论
TBI是神经外科的常见病、多发病,约占全身各处损伤的10%~15%。临床上拥有极高的致残、致死率。尤其是在战创伤中颅脑损伤所致阵亡比例居首[6]。在美国每年因TBI而致死、致残人数高达50,000和90,000人。在欧盟TBI的发生率高达100~800例/10万人,颅脑损伤每年入院人数超过100万例[7-9]。我国在上世纪80年代初TBI病死率高达10%左右[10-11]。目前随着社会发展,交通和生产事故不断增加,TBI在我国的发病率也呈不断上升的趋势。所以,TBI后神经细胞的损伤机制值得我们去深入研究。

图3 神经元损伤后流式细胞术检测

图4 谷氨酸模型中RIP1/3干扰质粒转染神经元存活情况

图5 炎症因子TNF-α诱导凋亡模型中RIP1/3干扰质粒转染神经元存活情况
TBI后脑组织主要的病理变化就是神经元和胶质细胞的坏死和凋亡。Rink等人于1995年发现,在大鼠颅脑外伤模型中大脑皮质、白质区,海马组织、齿状回等结构中出现了大量包括神经元等的凋亡现象[12]。Conti等人发现在TBI后1和7 d分别存在2个不同的凋亡高峰,并且星形胶质细胞(astrocyte)和少突胶质细胞(oligodendroglia)等神经胶质细胞也参与到了神经元的凋亡活动中[13]。Williams等的研究表明,在TBI 1年后大脑白质区内仍能明显地发现神经细胞凋亡[14]。
创伤后短时间内造成的神经元大量死亡多为直接损伤因素所导致的坏死,随后细胞凋亡与坏死则同时存在于创伤区的组织中。TBI后大脑的血液循环受到影响,导致脑组织发生缺血缺氧反应,神经元与胶质细胞的生物膜功能出现障碍。这些病理反应反过来会使血管和细胞毒性的脑水肿发生,进一步加重了脑损伤。缺血与缺氧环境下亦可造成神经元的能量代谢失衡与各类兴奋性氨基酸的过量释放。神经元内Ca2+失衡,脂质、过氧化物和氧化自由基等也会发生变化。以上神经细胞微环境的改变促使调控细胞凋亡的基因如bcl-2和P53等表达上调或下调,激活Caspase及RIP蛋白家族等[15],最终启动了细胞的自杀程序。
2005年Degterevs关于缺血性脑损伤模型的细胞死亡机制探讨中发现了necrostatin-1(Nec-1)。Nec-1是一种生物小分子,它可以抑制Fas/TNFR l的一种下游通路,即受体相互作用蛋白1(recepter interacting protein 1,RIP 1)所介导的细胞坏死,但却不能抑制Fas/TNFR l依赖性的细胞凋亡。说明存在某种也可以受相应的分子调控的细胞坏死。由此一条可介导细胞程序性坏死的调控分子被发现了,而它被证明位于DD下游的信号通路[16]。Nec-l则被认为是这种程序性坏死的特异性阻断剂,广泛使用在相关实验中。Nec-l作用于RIP 1与RlP 3的丝/苏氨酸激酶活性位点,抑制了RIP 1与RIP 3的相互磷酸化。进一步阻碍了RIP1-RIP3-TNFR复合体II的形成,从而抑制细胞坏死[17]。在谷氨酸诱导的海马神经元坏死[18]、小鼠大脑皮层神经元的缺血-再损伤灌注[19]、视网膜缺血损伤[20]以及阿尔茨海默病(Alzheimer's disease)、帕金森氏综合征(Parkinson's disease)和亨廷顿氏舞蹈病(Huntington's disease)等多种神经疾病中都证实存在这种RIP1-RIP3蛋白所影响的程序性坏死[21-23]。
本实验研究显示,不论是干扰RIP1或RIP3都对TBI后神经元具有明显的保护作用,其保护机制尚不清楚,可能通过多种途径影响神经细胞损伤后凋亡、坏死、自噬与存活;同时RIP1的抑制剂Nec-1也具有神经保护作用,进一步说明受体相互作用蛋白在TBI后调节神经元死亡具有重要作用。Nec-1对于神经元的保护作用比单纯干扰RIP蛋白更强,提示Nec-1可能还存在着其他神经保护作用。
谷氨酸是大脑皮层内最具代表性的兴奋性神经递质;它与神经元的发育、死亡、信息传递和突触的可塑性以及神经系统疾病密切相关。TBI后谷氨酸大量释放产生神经毒性,在神经系统损伤中发挥着独特作用,是其他系统损伤所少有的。一直以来Necrostatin-1都被认为是RIP1/3蛋白磷酸化的特异性抑制剂[24]。其作为特异性的程序性坏死抑制剂而被广泛应用。Xu等发现nec-1在HT-22海马神经元系中可以抑制谷氨酸引起神经兴奋毒性[19]。也有实验证实Nec-1对于脑缺血模型具有保护作用。但是通过上述实验可以看出RIP1/3蛋白对于谷氨酸诱导的神经细胞死亡不具有保护作用,因此nec-1对于谷氨酸神经兴奋毒性的保护靶点可能不是RIP1/3。这与之前报道的推断存在矛盾。同时干涉RIP1/3并未降低谷氨酸细胞毒性,也说明RIP1/3可能不是神经兴奋性毒性的下游作用分子(图6)。王玉田院士研究小组发现谷氨酸对GABA受体有增强变构作用,研究发现神经元损伤后释放大量谷氨酸达到一定阈值时谷氨酸会与抑制性GABA受体结合,产生神经抑制效果,有益于保护神经元生存[25]。Nec-1对谷氨酸损伤性神经元模型的保护作用,是否是其促进谷氨酸与GABA受体结合导致,有待进一步系统研究。

图6 Nec-1对TBI后不同细胞凋亡通路的影响模型
TBI后脑组织尤其是神经元的反应十分复杂,有研究认为最初的创伤可能导致损伤级联效应,引发多种病理生理过程。本研究证实RIP1与RIP3所诱导的细胞凋亡或程序性坏死也参与其中,并且这种死亡方式与谷氨酸的兴奋性神经毒性无关,而RIP1的抑制剂necrostatin-1对于谷氨酸损伤的保护作用提示其不仅仅是RIP蛋白的特异性抑制剂,其在TBI中可能存在其他的作用靶点。以上研究结果为进一步探究TBI后细胞分子生物学机制提供了一些参考。在这一方面的后续研究将对于临床开发创伤性颅脑损伤防治药物具有重要意义。
[1] Moretti L,Cristofori I,Weaver SM,et al.Cognitive decline in older adults with a history of traumatic brain injury[J].The Lancet Neurology,2012,11(12):1103-1112.
[2] Langlois JA,Rutland-Brown W,Wald MM.The epidemiology and impact of traumatic brain injury:a brief overview[J].J Head Trauma Rehabil,2006,21(5):375-378.
[3] Graham DI,Mcintosh TK,Maxwell WL,et al.Recent advaces in neurotrauma[J].J Neuropathol Exp Neurol,2000,59(8):641-651.
[4] Ai FD.O'leary DM,fan L,Bao W,mullins PG,movsesyan VA[J].Selective blockade of the mGluR1 trauma.Exp Neurol,2001,167(2):435-444.
[5] Huang WD,Fei Z,Zhang X.Traumatic injury induced homer-1a gene expression in cultured cortical neurons of rat[J].Neurosci Lett,2005,389(1):46-50.
[6] Rosonke S,Legome E.Head trauma[J].J emerg Med,2006,31(4):421-425.
[7] 费舟.现代颅脑损伤学[M].北京:人民军医出版社,2007.
[8] Federal Interagency Head Injury Task Force Report.Washington[Z],1989.
[9] Langlois JA,Rutland-Brown W,Tomas K.Traumatic brain injury in the United States:emergency department visits hospitalization and deaths[Z],2004.
[10]Thurman DJ,Alverson C,Dunn KA,et al.Traumatic brain injury in the United States:A public health perspective[J].J Head Trauma Rehabil,1999,14(6):602-615.
[11]Zhao YD,Wang W.Neurosurgical trauma in People's Republic of China[J].World J Surg,2001,25(9):1202-1204.
[12]Rink A,Fung KM,Trojanowski JQ,et al.Evidence of apoptosis cell death after experience traumatic brain injury in rat[J].Am J Pathol,1995,147(6):1575.
[13]Conti AC,Raghupathi R,Trojanowski JQ,et al.Experimental brain injury induces regionally distinct apoptosis during the acute and delayed post-traumatic period[J].J Neurosci,1998,18(15):5663-5672.
[14]Williams S,Raghupathi R,Mackinnon MA,et al.In situ DNA fragmentation occurs in white matter up to 12 months after head injury in man[J].Acta Neuropathol,2001,102(6):581-590.
[15]雪亮,杨树源.人脑创伤后神经元凋亡及调节机制的观察[J].中华创伤杂志,2003,19(3):160-163.
[16]Degterev A,Huang ZH,Boyce M,et al.Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury[J].Nat Chem Biol,2005,1(2):112-119.
[17]Han WD,Xie JS,Li L,et al.Necrostatin-1 reverts shikonin-induced necroptosis to apoptosis[J].Apoptosis,2009,14(5):674-686.
[18]Xu X,Chua CC,Kong J,et al.Necrostatin-1 protects against glutamate-induced glutathione depletion and caspase-independent cell death in HT-22 cells[J].J Neurochem,2007,103(5):2004-2014.
[19]Xu X,Chua KW,Chua CC,et al.Synergistic protective effects of humanin and necrostatin-1 on hypoxia and ischemia/reperfusion injury[J].Brain Res,2010,1355(2):189-194.
[20]Rosenbaum DM,Degterev A,David J,et al.Necroptosis,a novel form of caspase-independent cell death,contributes to neuronal damage in a retinal ischemia-reperfusion injury model[J].J Neurosci Res,2010,88(7):1569-1576.
[21]Lin MT,Beal MF.Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases[J].Nature,2006,443(7113):787-795.
[22]Li Y,Yang X,Ma C,et al.Necroptosis contributes to the NMDA-induced excitotoxicity in rat's cultured cortical neurons[J].Neurosci Lett,2008,447(2/3):120-123.
[23]Zhu S,Zhang Y,Bai G,et al.Necrostatin-1 ameliorates symptoms in R6/2 transgenic mouse model of Huntington's disease[J].Cell Death Dis,2011,2(1):e115.
[24]Pascual MJ,Reeds PJ.In vivo glucose contribution to glutamate synthesis is maintained while its contribution to acetyl CoA is lowered in adult mice fed a restricted amount of carbohydrate J[J].Nutr,1998,128(128):733-739.
[25]Liu J,Wu DC,Wang YT.Allosteric potentiation of glycine receptor chloride currents by glutamate[J].Nat Neurosci,2010,13(10):1225-1232.
(2016-07-17收稿)
The effect and mechanism of RIP1/3 on neurons after traumatic brain injury
WangHongbin,ZhengXinrui,YuanHua,etal.
DepartmentofRehabilitationandPhysiotherapy,XijingHospital,TheFourthMilitaryMedicalUniversity,Xi’an710032
Objective To investigate the effect and mechanism of RIP1/3 on neurons after traumatic brain injury (TBI).Methods The cultured cortical neurons were divided into four groups:siRIP group,Nec-1 pretreatment group,negative plasmid transfection group and control group.The TBI damage model was established by interfering with the expression of RIP1 and RIP3 through lentiviral transfection,respectively.The RIP3 damage model was established,and the survival rate of neurons was detected by flow cytometry.Results After mechanical injury,the survival rate of neurons in the siRIP group and in the Nec-1 pretreatment group was higher than that in the negative plasmid transfection group and in the control group.After glutamate stimulation,the survival rate of neurons in the Nec-1 pretreatment group was higher than that in the control group.siRIP3 group or siRIP1 group showed no significant difference in the survival rate of neurons when contrasted with the negative control transfection group.Conclusion RIP1 and RIP3 may play a role in neuronal death induced by TBI,RIP1 inhibitor Nec-1 may have a function in brain protection for TBI.RIP1 and RIP3 have no effect on neuron cell death induced by glutamate excitotoxicity.Nec-1 has a potential protective effect on glutamate injury,probably suggesting a specific target.
Traumatic brain injury RIP1/3 Neuron Glutamate
国家科技部国际合作专项项目(编号2013DFA32610);国家自然科学基金资助项目(81271450);陕西省国际科技合作与交流计划项目(2015KW-035)
710032 西安,第四军医大学西京医院康复理疗科[王宏斌(共同第一作者,硕士在读) 袁华(通讯作者)],神经外科 [郑新瑞(共同第一作者)]; 西安交通大学医学部研究生院康复与理疗专业[何芳梅]
R741
A
1007-0478(2017)01-0045-06
10.3969/j.issn.1007-0478.2017.01.012
