长期误诊的肉芽肿性皮肤松弛症1例
2017-07-31董丽儒宋旭东
董丽儒,张 莹,李 双,宋旭东
长期误诊的肉芽肿性皮肤松弛症1例
董丽儒1,张 莹2,李 双1,宋旭东1
肉芽肿性皮肤松弛症;蕈样霉菌病;误诊
患者女性,26岁,主因发现皮肤瘙痒及右下腹出现包块半年余入院就诊,查体发现皮损大小15 cm×9 cm,边界不清,质地硬,无触痛。局部切除后数月切口迁延不愈。2年余腹部、面部皮肤出现松弛、膨大、下垂,皮肤呈暗红色,症状渐进性加重(图1)。
病理检查 眼观:带皮肤组织3块,总大小7 cm×5 cm×5 cm,切面灰白、灰红色,质实,稍脆,边界模糊不清,表面皮肤局灶有糜烂。镜检:真皮及皮下组织内见大量中~小型淋巴细胞浸润(图2),伴多核巨细胞反应及淋巴吞噬现象(图3);表皮内及真表皮交界处见伴空晕的灶状淋巴细胞浸润,局灶伴浅表糜烂。大部分皮肤附属器消失,见神经纤维、血管壁、汗腺导管浸润现象。淋巴细胞未见明显核异型,未见核分裂象。弹性纤维染色:真皮弹力纤维断裂、显著减少甚至消失(图4)。免疫表型:CD3、CD2、CD4(图5)、CD68和CD5均呈阳性,CD7部分呈阳性,CD8呈散在阳性,CD20、CD23、CD10、CD30、ALK、BCL-2、CD56、MPO、TIA-1、GranzymeB、Perforin、Cyclin D1均呈阴性,Ki-67增殖指数为3%。
病理诊断:右下腹肉芽肿性皮肤松弛症(granulomatous slack skin, GSS)。患者随访4年半,因多脏器功能衰竭死亡。
讨论 GSS是一种极为罕见的嗜表皮性的原发性皮肤T细胞淋巴瘤,特点是在较大皮肤皱褶部位(腋下、腹股沟)出现缓慢发展的皮肤松弛,并可继发溃疡[1]。多数GSS患者临床经过呈惰性,Noto等[2]报道1/3患者与经典型霍奇金淋巴瘤有关,van Haselen等[3]报道与典型的蕈样霉菌病有关。GSS于1973年由Convit等[4]首先报道,Ackerman于1978年提出GSS这一诊断;国内最先由陈明华等[5]报道。WHO(2008)造血与淋巴组织分类明确提出GSS是蕈样霉菌病的一个亚型,临床极为罕见,相比其他亚型预后较差[6]。该病误诊极多,因其起病隐匿,早期临床表现不特异,常表现为硬斑,界限清楚,皮下组织肿胀,患者无特别不适,早期往往被忽视或被误诊为其他疾病[7]。
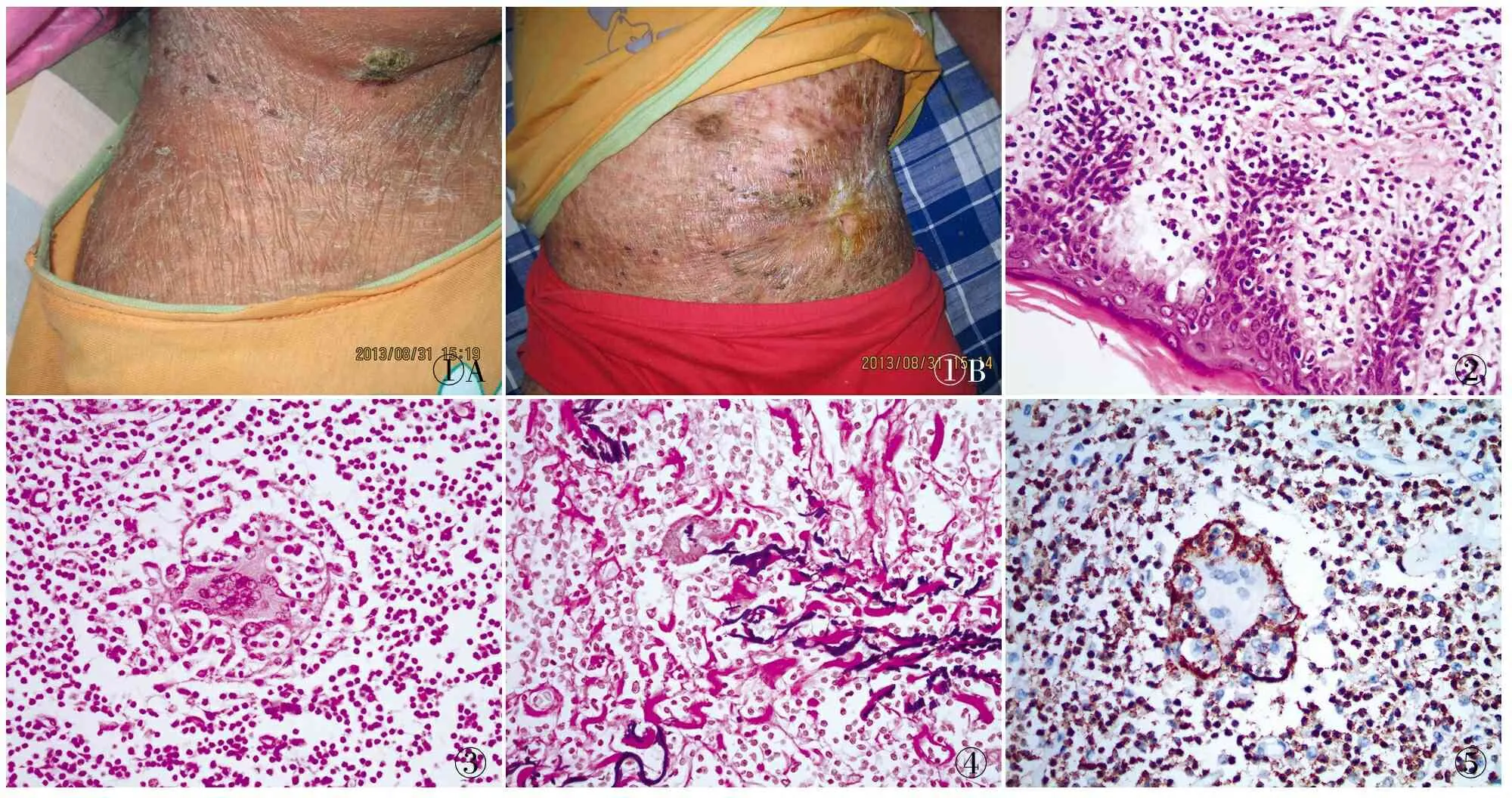
①A①B②③④⑤
图1 患者疾病晚期右颈部及腹部可见暗红色斑块及溃疡:A.颈部;B.腹部 图2 真皮可见单个核细胞浸润,细胞核型不规则,并可见侵犯表皮 图3 单个核细胞密集,可见多核巨细胞吞噬现象 图4 多核巨细胞吞噬弹力纤维,弹力纤维染色 图5 单个核细胞CD4呈阳性,SP法
GSS的诊断主要依赖于其临床表现、形态学表现、免疫表型,有时甚至需要TCR基因重排来确诊。本病呈慢性迁延过程,大多数患者具有惰性临床过程,患者早期并不会出现典型的皮肤松弛症状,常在发病数年后出现皮肤松弛,表现为皮肤弹性降低、起皱、悬垂及萎缩等,皮肤松弛可局限于某一部位,也可以广泛发于全身,因此该类疾病的早期诊断主要依赖于病理活检[8]。GSS组织学表现为皮下克隆性非典型性淋巴细胞、大量组织细胞和多核巨细胞形成的肉芽肿性浸润,并可见多核细胞吞噬弹力纤维及淋巴样细胞[9]。克隆性T细胞除表达CD2、CD3、CD4和CD5等T细胞抗原外,还会表达TIA-1,罕见表达CD8[10];不同病例中具有T细胞受体基因克隆性重排[11]。
本例首先以皮肤瘙痒及皮下肿块起病,切除肿块后形成迁延不愈的皮肤溃疡,当时其并未出现典型的皮肤松弛症状,经数年后全身以皱褶部位为主出现斑块,并发展成为典型的松弛表现,最终经病理检查明确诊断。患者因皮肤经久不愈的溃疡再次就诊时出现明显的皮肤松弛的临床表现,进一步复习患者2年前切片发现真皮及皮下组织内见大量中、小型淋巴细胞浸润,伴多核巨细胞反应及淋巴吞噬现象;表皮内及真表皮交界处见伴空晕的灶状淋巴细胞浸润,淋巴细胞未见明显核异型,未见核分裂象,局灶伴浅表糜烂,大部分皮肤附属器消失,可见神经纤维、血管壁、汗腺导管浸润现象。由于本病罕见且患者初发病是皮损,临床表现无特异性,另患者当时并未出现典型的皮肤松弛症状,故当时病理诊断报告肉芽肿性炎,而未进行免疫组化及特殊染色等检查,导致患者长期无法确诊。该病目前无特效疗法,手术切除皮损数月内可复发,用肾上腺皮质激素治疗、化疗或干扰素治疗,可获得部分缓解、延缓病程[12]。该病早期不易诊断,做病理检查可以明确诊断,其发展缓慢,但继发恶性淋巴瘤的危险性高(约50%),常见合并霍奇金淋巴瘤,故应加强随访。
[1] 闫 娜,刘栋华. 肉芽肿性皮肤松弛症1例[J]. 中国皮肤性病学杂志, 2014,28(8):829-832.
[2] Noto G, Pravatà G, Miceli S, Aricò M. Granulomatous slack skin: report of a case associated with Hodgkin’s disease and a review of the literature[J]. Br J Dermatol, 1994,131(2):275-279.
[3] van Haselen C W, Toonstra J, van der Putte S J,etal. Granulomatous slack skin. Report of three patients with an updated review of the literature[J]. Dermatology,1998,196(4):382-391.
[4] Convit J, Kerdel F, Goihman M, Rondon A J,etal. Progressive, atrophying, chronic granulomatous dermohypodermitis. Autoimmune disease[J]? Arch Dermatol, 1973,107(2):271-274.
[5] 陈明华,邱丙森,孔今城,等. 肉芽肿性松弛皮肤特殊类型的蕈样肉芽肿一例[J]. 中华皮肤科杂志, 1999,32(6):370-372.
[6] Swerdloe S H, Campo E, Harris N L,etal. WHO classification of tumors of haematopoietic and lymphoid tissues[M]. Lyon: IARC Press, 2008:535-541.
[7] Goldsztajn K O, Moritz Trope B, Ribeiro Lenzi M E,etal. Granulomatous slack skin. Histopathology diagnosis preceding clinical manifestations by 12 years[J]. J Dermatol Case Rep, 2012,6(4):108-112.
[8] Kogut M, Hadaschik E, Grabbe S,etal. Granulomatous mycosis fungoides, a rare subtype of cutaneous T-cell lymphoma[J]. JAAD Case Rep, 2015,1(5):298-302.
[9] Wieser I, Wohlmuth C, Duvic M. Granulomatous mycosis fungoides in an adolescent-A rare encounter and review of the literature[J]. Pediatr Dermatol, 2016,33(5):e296-e298.
[10] Ishida M, Hotta M, akikita-Suzuki M,etal. CD8-positive granulomatous mycosis fungoides: a case report with review of the literature[J]. J Cutan Pathol, 2010,37(10):1072-1076.
[11] Dabiri S, Morales A, Ma L, Sundram U,etal. The frequency of dual TCR-PCR clonality in granulomatous disorders[J]. J Cutan Pathol, 2011,38(9):704-709.
[12] Wollina U, Graefe T, Füller J. Granulomatous slack skin or granulomatous mycosis fungoides-a case report. Complete response to percutaneous radiation and interferon alpha[J]. J Cancer Res Clin Oncol, 2002,128(1):50-54.
华北理工大学附属医院1病理科、2重症医学科,唐山 063000
董丽儒,女,博士研究生,主治医师。E-mail:470818292@qq.com 宋旭东,男,硕士生导师,主任医师,通讯作者。E-mail:songxd2002@sina.com
时间:2017-6-20 11:19 网络出版地址:http://kns.cnki.net/kcms/detail/34.1073.R.20170620.1117.033.html
R 739.5
B
1001-7399(2017)06-0707-02
10.13315/j.cnki.cjcep.2017.06.033
接受日期:2017-03-02
