高效液相色谱-串联质谱法测定虾中万古霉素及去甲万古霉素残留量
2017-06-29薛婷婷黄冬梅严敏鸣史永富田良良
薛婷婷,黄冬梅,严敏鸣,韩 峰,王 政,史永富,田良良
(1.中国水产科学研究院 东海水产研究所,上海 200090;2.上海海洋大学 食品科学与工程,上海 201306; 2.上海市浦东新区农产品检测中心,上海 201202)
高效液相色谱-串联质谱法测定虾中万古霉素及去甲万古霉素残留量
薛婷婷1,2,黄冬梅1*,严敏鸣3,韩 峰1,王 政3,史永富1,田良良1
(1.中国水产科学研究院 东海水产研究所,上海 200090;2.上海海洋大学 食品科学与工程,上海 201306; 2.上海市浦东新区农产品检测中心,上海 201202)
建立了虾中万古霉素和去甲万古霉素残留量的高效液相色谱-串联质谱(HPLC-MS/MS)检测方法。样品经0.1%甲酸-乙腈(9∶1,体积比)混合溶液提取,乙腈饱和的正己烷除脂,PCX结合Florisil固相萃取柱进行净化后,以乙腈和0.1%甲酸溶液为流动相,经CAPCELL PAK MG-C18色谱柱分离后,采用高效液相色谱-串联质谱在多反应离子监测(MRM)模式进行测定,以双去氯万古霉素作为内标物,内标法定量。结果表明:万古霉素和去甲万古霉素在2~250 ng/mL范围内呈良好的线性关系,相关系数(r2)均大于0.99。在5,25,50 μg/kg加标水平下,万古霉素和去甲万古霉素的平均回收率为87.2%~102%,相对标准偏差为1.3%~8.7%。方法的检出限(LOD)为2.0 μg/kg,定量下限(LOQ)为5.0 μg/kg。该方法灵敏、准确,重复性好,适用于虾类中万古霉素及去甲万古霉素残留量的测定。
万古霉素;去甲万古霉素;双去氯万古霉素;虾;高效液相色谱-串联质谱(HPLC-MS/MS)
万古霉素和去甲万古霉素是由东方链霉菌代谢产生的糖肽类抗生素[1]。这种药物通过抑制细胞壁中磷脂和多肽的生成,进而干扰细胞壁的合成[2],对革兰阳性球菌与杆菌具有强大抗菌作用[3]。此外,万古霉素因其特殊的分子结构,可有效治疗一些耐药菌株,如超级病菌NDM-1等[4]。因此万古霉素和去甲万古霉素作为抗生素被不法经营者用于养殖业的情况日渐增多[5]。该类药物可通过食物链进入人体,导致人类听力减退甚至缺失,肾小管受损,甚至造成肾衰竭[6]。一些国家制定了相关食品中的残留限量标准[7],日本规定牛奶中万古霉素的限量值为10 μg/kg[8]。而我国农业部第560号公告中规定万古霉素为禁用兽药[9],未提出万古霉素在食品中的最大残留限量MRL值。卫生部将万古霉素列入《食品中可能违法添加的非食用物质和易滥用的食品添加剂名单(第1-5批汇总)》,并注明需研制针对动物性食品中该药物的检测方法[10]。目前虽有猪肉[5]和乳制品[6]中万古霉素残留量的相关检测文献,但水产品领域中的相关研究尚属空白。我国是虾类主要养殖大国,虾产品出口贸易呈逐年增长趋势,因此,建立一种适用于虾类中万古霉素和去甲万古霉素残留量的检测方法具有重要的意义。
国内外已有报道的万古霉素等糖肽抗生素含量的主要检测方法有免疫法[11-16]、高效液相色谱法(HPLC)[17-20]、液相色谱-串联质谱法(HPLC-MS/MS)[21-24]。对于基质复杂的样品,免疫法因专属性较差,所以较少采用。相对HPLC法,HPLC-MS/MS具有灵敏度高、定性更准确的优势。而选用内标法定量,可有效降低实际操作中条件波动对分析结果的影响。本文选择双去氯万古霉素作为内标物,利用高效液相色谱-串联三重四极杆质谱仪测定虾类中万古霉素及去甲万古霉素,该方法具有较好的检测准确性和灵敏度,可满足残留检测的要求。
1 实验部分
1.1 仪器与试剂
Ultimate 3000高效液相色谱-串联TSQ ENDURA三重四极杆质谱(美国Thermo Fisher公司);Genius 3旋涡混合器(德国IKA公司);5510超声波清洗仪(美国Branson公司);Multifuge X 3R高速离心机(Thermo公司);BCD-215DC冰箱(中国Haier公司);N-EVAPTM111型氮吹仪(美国Organomation Aossociates公司);VisiprepTMDL固相萃取装置(美国Supelco公司);PCX固相萃取柱(200 mg/6 mL,Aglient公司);Florisil固相萃取柱(500 mg/6 mL,Agela公司)。
万古霉素(纯度94.1%,Dr.Ehrenstorfer GmbH);去甲万古霉素(纯度83.4%,中国药品生物制品检定所);双去氯万古霉素(纯度75%,Toronto Research Chemicals);甲醇、乙腈(HPLC级,J,T.Baker);甲酸(分析纯,国药集团化学试剂有限公司);正己烷(HPLC级,J.T.Baker);0.1%甲酸溶液-乙腈混合溶液(7∶3,体积比);5%氨化甲醇。
1.2 标准溶液的配制
混合标准储备液:分别准确称取万古霉素、去甲万古霉素和双去氯万古霉素,用0.1%甲酸溶解,配制成100 μg/mL的标准混合储备液和内标液;再分别用0.1%甲酸稀释成1 μg/mL混合标准溶液和内标液。根据需要,用0.1%甲酸溶液逐级稀释成2,5,10,25,50,100,250 ng/mL的标准工作液,其中内标液浓度为50 ng/mL,现用现配。以上标准溶液于4 ℃冰箱避光冷藏。
1.3 样品前处理
1.3.1 提 取 准确称取虾样品2 g(精确至0.01 g)于50 mL聚丙烯离心管中,加入100 μL 1 μg/mL的双去氯万古霉素内标液,用9 mL 0.1%的甲酸-乙腈混合溶液提取,涡旋混合1 min,超声10 min,8 000 r/min离心6 min,取上清液至另一50 mL离心管中。残渣重复提取1次,合并上清液,用提取液定容至20 mL,取10 mL待净化。
1.3.2 净 化 在上述溶液中加入5 mL乙腈饱和的正己烷,充分振荡混匀,4 000 r/min离心10 min,去除上层正己烷层,重复去脂1次,下层溶液备用过柱。PCX固相萃取柱依次用5 mL乙腈和5 mL 0.1%甲酸溶液活化,Florisil固相萃取柱依次用5 mL甲醇和5 mL 5%氨化甲醇活化。取溶液以每秒1滴的速度过PCX柱,10 mL蒸馏水淋洗,抽干柱子,5 mL 5%氨化甲醇洗脱,收集洗脱液过Florisil柱,收集过柱液,3 mL 5%氨化甲醇淋洗,收集全部淋洗液,于45 ℃氮气吹至近干。以1 mL 0.1%甲酸复溶,混匀,过0.22 μm滤膜后,供 LC-MS/MS分析。
1.4 色谱条件
色谱柱:CAPCELL PAK MG-C18(100 mm×2.0 mm,5 μm);流速:0.3 mL/min;柱温:30 ℃;进样量:25 μL;流动相:A为0.1%甲酸,B为乙腈。洗脱梯度:0~0.5 min,95% A;0.5~2.5 min,95%~75% A;2.5~5.0 min,75% A;5.0~6.1 min,75%~95% A;6.1~8.1 min,95% A。

表1 万古霉素、去甲万古霉素及双去氯万古霉素的质谱参数
*quantitative ion
1.5 质谱条件
离子源:电喷雾离子源(ESI);检测方式:多反应离子监测(MRM);扫描方式:正离子模式;喷雾电压:3 000 V;鞘气流量:35 mL/min;辅助气流量:10 mL/min;离子传输毛细管温度:350 ℃。其他质谱参数见表1。
2 结果与讨论
2.1 质谱条件的优化
万古霉素、去甲万古霉素和双去氯万古霉素均为多肽类抗生素,在一级质谱中,易与H+结合,产生带正电的双电荷离子[M+H]2+,经过二级碰撞后,产生不同强度的碎片离子,以丰度最大的m/z144.0,144.0,100.1分别作为3种化合物的定量离子。确定特征碎片离子后,对碰撞能量等其他质谱参数进行优化,优化后的质谱参数见“1.5”及表1。
2.2 色谱条件的选择
因甲酸可为化合物提供必需的质子来源,提高其离子化效率[5],故在万古霉素和去甲万古霉素残留分析中使用的流动相多为0.1%甲酸-乙腈体系。经查阅,万古霉素在C18柱上有较好的保留效果[1],本文比较了AgilentEC-C18(2.1 mm×100 mm,2.7 μm),AgilentGlycan-C18(2.1 mm×100 mm,1.8 μm)和Capcell PAK MG-C18(2.0 mm×100 mm,5 μm) 3种不同C18色谱柱的效果,结果显示Capcell PAK MG-C18色谱柱对万古霉素和去甲万古霉素的保留效果最好。在此条件下万古霉素、去甲万古霉素及双去氯万古霉素的响应值高,且峰形尖锐、对称。
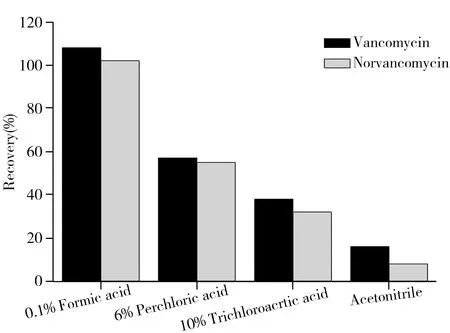
图1 4种不同提取溶液对万古霉素和去甲万古霉素的提取效果比较Fig.1 Comparison recoveries of vancomycin and norvancomycin by four different kinds of extract solutions
2.3 前处理条件的优化
2.3.1 提取试剂的选择 根据万古霉素和去甲万古霉素易溶于水,微溶于乙腈的性质,本文比较了0.1%甲酸、10%三氯乙酸、6%高氯酸以及纯乙腈4种试剂作为提取溶液时对加标回收率的影响,结果见图1。纯乙腈的提取回收率最低,三氯乙酸和高氯酸的提取回收率仅为40%~60%,而0.1%甲酸的提取回收率最高,但会有水溶性蛋白溶出,造成杂质较多,提取液较为浑浊,不利于下步净化。因此本文在0.1%甲酸中加入一定比例的乙腈,采用0.1%甲酸-乙腈(7∶3,体积比)提取,该条件下提取回收率高,同时能沉淀部分蛋白和杂质,利于下步净化。
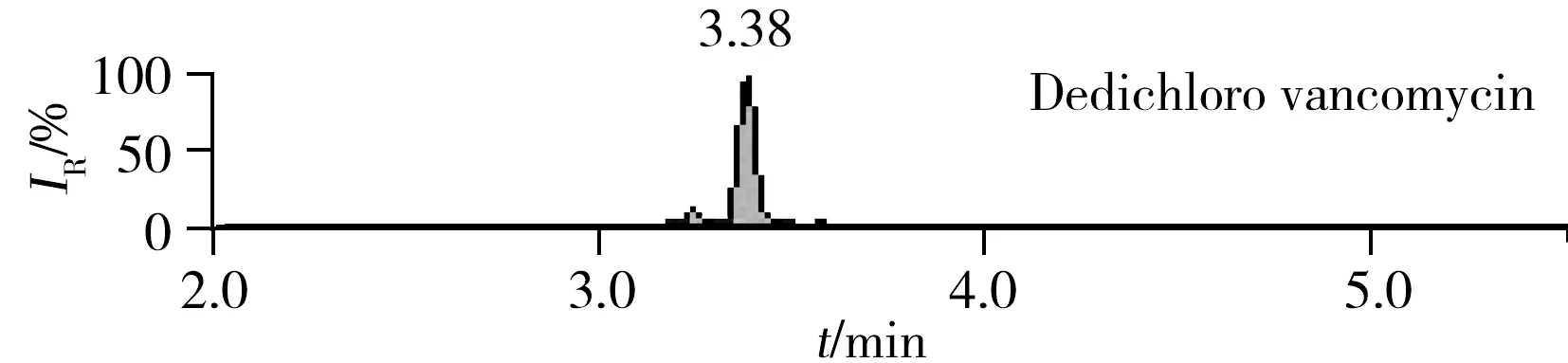

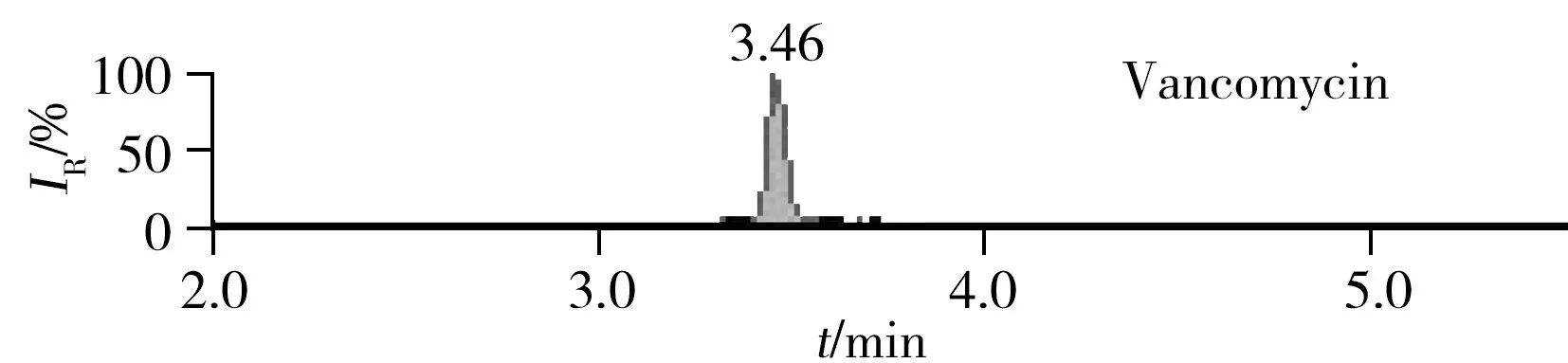
图2 罗氏沼虾加标样品图(25 μg/kg)Fig.2 Spiked samples of Giant freshwater prawn(25 μg/kg)
2.3.2 净化及固相萃取小柱的优化 首先采用正己烷(乙腈饱和)去除提取液中的部分脂类杂质,而后采用固相萃取小柱进一步净化处理。万古霉素等目标物分子结构中含有1个碱性的伯氨基团,带有弱碱性,而虾类样品中富含色素等杂质,因此本实验采用以水浸润型聚合物为基质的阳离子交换柱PCX柱和硅胶键合氧化镁吸附剂的Florisil固相萃取柱联合去杂,结果表明此方法能够有效去除杂质,具有良好的净化效果,且两种化合物的回收率均在80%~110%之间,能满足检测要求。 阴性罗氏沼虾样品中添加25 μg/kg目标物的加标谱图见图2。
2.4 内标物的选择
目前已报道[25]用于万古霉素和去甲万古霉素残留的内标物有甲硝唑和维生素B12,但其在实际样品前处理过程中损失较大,不能满足本实验作为内标物的要求。作者通过万古霉素相关性质资料查询[26],发现双去氯万古霉素的结构和基本性质与万古霉素相似,满足内标物的要求。因此,本文采用双去氯万古霉素作为内标物,使用该内标物进行复杂基质样品中万古霉素和去甲万古霉素检测的研究尚未见报道。
2.5 线性范围、检出限与定量下限
将浓度分别为2,5,10,25,50,100,250 ng/mL的待测物系列混合标准工作液上机测定,以目标物和内标物的峰面积比值(y)为纵坐标,标准溶液浓度(x,ng/mL)为横坐标,绘制标准曲线。结果显示,万古霉素和去甲万古霉素在2~250 ng/mL范围内线性关系良好,相关系数(r2)均大于0.99,其线性范围、线性方程、相关系数见表2。在虾空白样品中添加2种目标化合物,按样品前处理步骤和仪器条件进行检测,根据信号响应值,以3倍信噪比确定方法检出限(LOD,S/N≥3),10倍信噪比确定定量下限(LOQ,S/N≥10),测得万古霉素和去甲万古霉素的检出限均为2.0 μg/kg,定量下限均为5.0 μg/kg。本方法具有较高的灵敏度。

表2 万古霉素和去甲万古霉素的线性范围、线性方程、相关系数、检出限及定量下限
2.6 回收率与相对标准偏差
选择不含待测目标物的罗氏沼虾空白基质样品进行加标回收实验,分别添加5,25,50 μg/kg 3个浓度水平的标准溶液,每个加标浓度做6个平行样,分别计算万古霉素和去甲万古霉素的回收率,测得万古霉素的平均添加回收率为91.8%~102%,去甲万古霉素为87.2%~98.4%,相对标准偏差(RSD)为1.3%~8.7%(见表3)。綦艳等[24]采用LC-MS/MS 检测乳制品中的万古霉素残留量,在15,60,150 μg/kg加标水平下的回收率约80%。数据表明,本文建立的方法准确度较高,具有较好的精密度。

表3 罗氏沼虾空白样品中添加2种目标物的平均回收率及相对标准偏差
2.7 实际样品的检测
从上海农贸市场购买南美白对虾、基围虾、青虾、河虾等多种虾类样品,在优化条件下进行检测,同时做质控样品。结果在上述样品中均未检出万古霉素和去甲万古霉素,质控样品中两种化合物的回收率为81.4%~106%,精密度小于10%,达到残留分析要求,数据准确可靠。同时由3家经过认证的实验室对本方法做验证实验,结果表明本方法满足残留检测的各项指标。
3 结 论
本文建立了高效液相色谱-串联质谱检测虾类样品中万古霉素和去甲万古霉素残留的分析方法,该方法首次使用双去氯万古霉素作为内标物进行定量分析。优化了样品前处理、色谱及质谱条件,有效提高了检测的灵敏度和准确度。该方法回收率高,定量下限和精密度满足残留检测的要求,适用于虾类中万古霉素及去甲万古霉素残留量的同时检测。
[1] Yan J Y,Guo Z M,Ding J J,Shen A J,Wang J X,Jin G W,Liang X M.Chin.J.Chromatogr.( 闫竞宇,郭志谋,丁俊杰,深爱金,王纪霞,金高娃,梁鑫淼.色谱),2015,33(9):951-956.
[2] Vila M M D C,De Oliveira R M,Goncalves M M.Quim.Nova,2007,30(2):395-399.
[3] Liu M,Hu C G.Chromatographia,2007,65(3/4):203-207.
[4] Zhang H,Fang Y,Li Y,Zhou T R.Chin.J.Pharm.Anal.(张虹,方昱,李英,周陶然.药物分析杂志),2008,28(4):591-594.
[5] Xian Y P,Chen L W,Luo H Y,Guo X D,Wu Y L,Luo D H,Hou X C.J.Instrum.Anal.(冼燕萍,陈立伟,罗海英,郭新东,吴玉銮,罗东辉,侯向昶.分析测试学报),2013,32(2):162-167.
[6] Guo Q L,Shi H L,Yang H M,Wang H,Liu Y Q.Sci.Technol.FoodInd.(郭启雷,史海良,杨红梅,王浩,刘艳琴.食品工业科技),2013,36(8):86-87,101.
[7] National Pharmacopoeia Committee.Pharmacopoeia of the People's Republic of China.Part 2.Beijing:China Medical Science Press(国家药典委员会.中华人民共和国药典.二部.北京:中国医药科技出版社),2010:636.
[8] Liu J J,Jin F,She Y X,Liu H B,Shi X M,Wang M,Wang J,Xu S Y.Chin.J.Anal.Chem.(刘佳佳,金芬,佘永新,刘洪斌,史晓梅,王淼,王静,徐思远.分析化学),2011,39(5):652-657.
[9] Ministry of Agriculture.No.560 Bulletin of the Ministry of Agricultrue of the People's Republic of China.(农业部.中华人民共和国农业部公告第560号).(2005-10-28)[2005-11-01].http://www.moa.gov.cn/zwllm/tzgg/gg/200511/t20051117_496523.htm.
[10] Ministry of Health of the People's Republic of China.Food May be Illegal and Non-food Substances Added to the List of Food Additives Have Been Abused(1-5 Batches Summary).(中华人民共和国卫生部.《食品中可能违法添加的非食用物质和易滥用物质和易滥用的食品添加剂名单(第1-5批汇总)》).(2011-04-19).[2011-04-22].http://www.moh.gov.cn/publicfiles/business/ht mLfiles/mohwsjdj/s9164/201104/51441.htm.
[11] Peng S W,Chen Y G,Zou J L.J.GuangdongPharm.College(彭斯维,陈永刚,邹吉利.广东药学院学报),2014,30(5):553-558.
[12] He F Y,Luo X Q,Zhang Y,Fu J Q,Cui Y T,Wu X S.HeilongjiangAnim.Husb.Vet.Med.(何方洋,罗晓琴,张禹,付军权,崔延婷,吴小胜.黑龙江畜牧兽医),2013,8:82-84.
[13] Wang W Z,Qian N P,Chen B,Ma Y.ChinPharm.(王维忠,钱南萍,陈斌,马莹.中国药业),2012,21(20):27-29.
[14] Haeseker M B,Croes S,Neef C,Bruggeman C A,Stolk L M,Verbon A.PlosOne,2014,9(11):e112008.
[15] Lin W W,Wu W,Jiao Z,Lin R F,Jiang C Z,Huang P F,Liu Y W,Wang C L.Eur.J.Clin.Pharmacol.,2016,72(1):29-37.
[16] Sym D,Smith C,Meenan G,Lehrer M.Ther.DrugMonit.,2001,23(4):441-444.
[17] Guo Z S,Wang H,Zhang Y S.J.Pharm.Biomed.,2005,41(10):310-314.
[18] Ding G S,Huang X J,Liu Y,Wang J D.Chromatographia,2004,59(7/8):443-449.
[19] Lin X L,Zhu J P,Fei Y.Chin.J.Clin.Pharm.(林秀丽,朱金平,费燕.中国临床药学杂志),2011,20(4):231-234.
[20] Li H.Chin.J.Med.Guide(李慧.中国医药导刊),2011,13(8):1454-1459.
[21] Sakamoto Y,Jinno Y,Shinodzuka I,Iwasaki Y,Ito R,Saito K.Anal.Sci.,2014,30(2):271-275.
[22] Lin W X,Sun X Q,Tian M,Yu L,Xiao S S,Li Z.Chin.DairyInd.(林维宣,孙兴权,田苗,于灵,肖珊珊,李哲.中国乳品工业),2009,3:46-48.
[23] Li H,Lian H,Xiao C Q,Li L.Chin.J.Clin.Pharm.(李贺,廉洪,肖昌钱,李力.中国临床药理学杂志),2014,8:721-723.
[24] Qi Y,Li J Q,Zhao M Q,Long S R,Li M H,She Z Y,Huang B Y.J.Instrum.Anal.(綦艳,李锦清,赵明桥,龙顺荣,郦明浩,佘之蕴,黄宝莹.分析测试学报),2013,32(6):768-771.
[25] Xu B,Li X,Zhang L.CentralSouthPharm.( 徐兵,李昕,张莉.中南药学),2009,2:100-102.
[26] Kirst H A,Thompson D G,Nicas T I.Antimicrob.AgentsChemother.,1998,42(5):1303-1304.
Determination of Vancomycin and Norvancomycin Residues in Shrimps by High Performance Liquid Chromatography-Tandem Mass Spectrometry
XUE Ting-ting1,2,HUANG Dong-mei1*,YAN Min-ming3,HAN Feng1,WANG Zheng3,SHI Yong-fu1,TIAN Liang-liang1
(1.East China Sea Fisheries Research Institute,Chinese Academy of Fishery Sciences,Shanghai 200090,China;2.College of Food Science and Technology,Shanghai Ocean University,Shanghai 201306,China;3.Testing Center of Agricultural Products,Pudong New Area,Shanghai 201202,China)
A new method for the determination of vancomycin and norvancomycin residues in shrimps by high performance liquid chromatography-tandem mass spectrometry(HPLC-MS/MS) was established.Samples were extracted with 0.1% formic acid-acetonitrile solution,defatted withn-hexane saturated with acetonitrile and purified with PCX and Florisil solid phase extraction column.The separation of two targets was carried out on a CAPCELL PAK MG-C18column by gradient elution with 0.1% formic acid-acetonitrile as mobile phase.The identification of two analytes was performed in multi-reaction monitoring(MRM),and the quantification was carried out by the internal standard method.Dedichloro vancomycin was used as an internal standard.The calibration curves showed good linearities in the range of 2-250 ng/mL with correlation coefficients(r2) greater than 0.99.The detection limits(LOD) of this method were both 2.0 μg/kg,and the limits of quantitation(LOQ) were 5.0 μg/kg.The recoveries of the target compounds at spiked levels of 5,25,50 μg/kg were in the range of 87.2%-102%,with relative standard deviations(RSDs) of 1.3%-8.7%.The results indicated that this method had high sensitivity,accuracy and good reproducibility.It was suitable for the simultaneous determination of ancomycin and norvancomycin residues in shrimps.
vancomycin;norvancomycin;dedichloro vancomycin;shrimps;high performance liquid chromatography-tandem mass spectrometry(HPLC-MS/MS)
2017-01-17;
2017-03-06
农业行业标准制定和修定农产品质量安全项目(2130109)
10.3969/j.issn.1004-4957.2017.06.012
O657.63;O629.5
A
1004-4957(2017)06-0773-05
*通讯作者:黄冬梅,研究员,研究方向:食品质量安全,Tel:021-65680121,E-mail:hdm2001@126.com
