微波酸提取/液相色谱-电感耦合等离子体质谱法测定动物源性中药中6种砷形态
2017-06-29刘成雁王志嘉吴志刚吴冬冬
曹 璨,刘成雁,王志嘉,吴志刚,崔 畅,吴冬冬
(辽宁省分析科学研究院,辽宁 沈阳 110015)
微波酸提取/液相色谱-电感耦合等离子体质谱法测定动物源性中药中6种砷形态
曹 璨*,刘成雁,王志嘉,吴志刚,崔 畅,吴冬冬
(辽宁省分析科学研究院,辽宁 沈阳 110015)
建立了微波酸提取/液相色谱-电感耦合等离子体质谱联用(LC-ICP-MS)测定动物源性中药中6种砷形态(亚砷酸盐As(Ⅲ),砷酸盐As(V),一甲基砷MMA,二甲基砷DMA,砷甜菜碱AsB和砷胆碱AsC)的分析方法。采用1% HNO3溶液在80 ℃微波提取10 min,经离心分层,过固相萃取SEP C18柱和0.45 μm滤膜,以25 mmol/L NH4H2PO4溶液(pH 6.7)-乙醇(99∶1,体积比)为流动相进行等度洗脱,各砷形态在10 min内实现基线分离。结果显示,6种砷形态在1.0~100.0 μg/L范围内线性关系良好,相关系数为0.999 5~0.999 7,方法检出限(LOD)为0.24~1.0 μg/kg,相对标准偏差(RSD)为0.96%~2.0%。将方法应用于地龙、水蛭、海螵蛸、桑螵蛸、石决明和鸡内金中6种砷形态的测定,加标回收率为94.2%~103.8%,提取效率为95.5%~102.8%,优于热提取法。方法快速、准确、重现性好,适用于动物源性中药及类似样品中的砷元素形态分析及质量监控。
微波酸提取;液相色谱-电感耦合等离子体质谱联用(LC-ICP-MS);动物源性中药;砷形态分析
传统动物源性中药有着显著的生物活性,可用于改善和调节人体的生理功能,在国内外受到广泛关注,但重金属元素超标问题使之应用受到限制[1]。砷元素作为重金属中的一种,药典中明确规定了其在部分中药中的限量[2],限量值为2~10 mg/kg,远超出国际标准。而不同形态的砷毒性差异很大,无机态的亚砷酸盐As(Ⅲ)和砷酸盐As(V)的毒性较高,一甲基砷(MMA)和二甲基砷(DMA)的毒性较小,而砷甜菜碱(AsB)、砷胆碱(AsC)和各类砷糖、砷脂几乎无毒[3],因而仅对砷总量限制不足以考察药物毒性,对砷形态进行分析是非常必要的。目前已有研究主要集中在食品、植物和环境分析方面,而对于动物源性中药的相关研究报道很少。
砷元素形态分析方法主要有液相色谱-原子荧光光谱联用(LC-AFS)法[4-5]和液相色谱-电感耦合等离子体质谱联用(LC-ICP-MS)法[6-7],但LC-AFS法目前只能分析4种砷形态,有一定局限性,而LC-ICP-MS能够分析8种以上砷形态[6],是目前常用的分析方法。中国药典提供了中药材中砷元素的形态分析方法[8],但该方法耗时长,批量分析效率有待改进。砷的提取常采用加热酸提取、超声甲醇/乙酸+水或流动相提取、微波水提取等[5,9-10],其提取效率各有不同,而微波酸提取法尚未见相关报道。
本研究采用微波酸提取/LC-ICP-MS联用法测定了地龙、水蛭、海螵蛸、桑螵蛸、石决明、鸡内金6种动物源性中药的6种砷形态(As(Ⅲ),As(V),MMA,DMA,AsB和AsC),从而为动物源性中药中砷元素形态分析提供了简便、快速、稳定的方法,可用于中药材的质量控制和监督。
1 实验部分
1.1 仪器与试剂
Agilent1260液相色谱仪,Agilent7700x电感耦合等离子体质谱仪,配有联用装置及MassHunter工作站(美国Agilent公司);PRP-X100阴离子色谱柱(4.1 mm×250 mm,10 μm,瑞士Hamilton公司);Mars6微波消解仪(美国CEM公司);KQ3200DE数控超声波清洗器(昆山超声仪器有限公司);CT14RD高速冷冻离心机(上海天美生化仪器有限公司);AE-240电子天平(瑞士Mettler Toledo公司);Milli-Q纯水仪(美国Millipore公司)。
砷形态溶液和总砷溶液标准物质(中国计量科学研究院):As(Ⅲ) GBW08666,As(V) GBW08667,MMA GBW08668,DMA GBW08669,AsB GBW08670,AsC GBW08671,砷GBW08611,浓度分别为(75.7±1.2) μg/g,(17.5±0.4) μg/g,(25.1±0.8) μg/g,(52.9±1.8) μg/g,(38.8±1.1) μg/g,(28.0±1.1) μg/g,1 000 μg/mL;硝酸、30%过氧化氢(优级纯,国药集团化学试剂有限公司);磷酸二氢钾、氨水(分析纯,国药集团化学试剂有限公司);乙醇(色谱纯,上海沃凯化学试剂公司)。调谐液10 μg/L(含Li,Y,Ce,Tl,Co;美国Agilent公司);所有标准溶液和样品的制备用水均为Milli-Q超纯水(电阻率>18 MΩ·cm)。
地龙、水蛭、海螵蛸、桑螵蛸、石决明、鸡内金由医院药剂科采购提供。
1.2 实验方法
1.2.1 质谱条件 等离子气流量15 L/min;载气流量0.7 L/min;补偿气流量0.3 L/min;RF功率1 550 W;采样深度8 mm;雾化室温度2 ℃;辅助气流量0.9 L/min;氧化物比率1.10%;双电荷比率1.09%;四极杆真空度1.1×10-4Pa ;QP偏转电压-2.8 V。
1.2.2 色谱条件 流动相:25 mmol/L NH4H2PO4溶液(氨水调至pH 6.7)-乙醇(99∶1);高压泵进样方式,等度洗脱;流速1.0 mL/min;进样量20 μL;压力上限40 MPa;柱温:室温;采集时间 10 min。
1.2.3 砷形态分析前处理 样品经高速粉碎机粉成粉末,过80目筛待测。
微波提取法:称取样品1 g于微波消解罐中,加入1% HNO3溶液20 mL,加塞。设定微波提取条件为温度80 ℃,功率600 W,升温时间3 min,保持时间7 min。
振荡热浸提法:称取样品1 g于50 mL离心管中,加入1% HNO3溶液20 mL,加塞但不密闭。于95 ℃水浴、低速振荡条件下提取2.5 h。
采用不同方法提取后,冷却至室温,配重后于高速冷冻离心机以8 000 r/min转速4 ℃下离心15 min,取上清液过SPE C18柱和0.45 μm有机滤膜。同时做试剂空白。
1.2.4 总砷前处理 称取样品0.5 g于微波消解罐中,加入5 mL HNO3,加盖静置过夜。加入2 mL H2O2,按微波消解程序(见表1)设定,反应结束后冷却至室温,于配套电热赶酸板上170 ℃赶酸至溶液剩余约1 mL,用水转移至100 mL容量瓶后,定容摇匀。同时做试剂空白。

表1 微波消解程序
1.2.5 提取态总砷前处理 分别移取10 mL“1.2.3”提取的待测液和空白溶液,按“1.2.4”方法处理。
2 结果与讨论
2.1 砷形态前处理条件的优化
2.1.1 提取试剂的选择 动物源性中药中含有蛋白、脂类等成分,若采用有机相+水提取,则脂类成分不易分离,增加了后期检测难度;而用水提取则效率较低;用流动相提取,受提取温度和pH值的限制,有一定的局限性,因此仅考虑稀酸溶液。由于盐酸中的35Cl在ICP-MS分析时与40Ar形成40Ar35Cl,会对75As产生质谱干扰;乙酸作提取试剂时含碳量较高,基线稳定性较差;而HNO3不引入干扰元素,是提取试剂首选。分别采用0.3%,1%,2%,5% HNO3溶液进行提取,结果表明增加酸度有利于提高提取效率,酸度达到1%时提取效率趋于稳定,2%时部分砷形态易发生转化,且酸度越大,砷形态转化越显著,因此选择1%HNO3溶液作为提取试剂。
2.1.2 提取方式的选择 分别用微波提取和振荡热浸提两种方式对地龙样品进行前处理,比较了各砷形态含量和提取态总砷含量。结果显示,微波提取法的提取率普遍略高于振荡热浸提法,且该方法提取温度低,提取时间大大缩短,因此选择微波浸提作为提取方式。
2.1.3 提取温度的选择 分别设定微波提取温度为70,80,90,100 ℃,比较了地龙样品中各砷形态含量和可提取态总砷的含量。结果表明70 ℃时可提取态总砷含量略低, 80 ℃后可提取态总砷含量基本保持不变,因此认为80 ℃时各形态砷已提取完全。当提取温度达100 ℃时,无机态砷的浓度明显增加,砷形态发生转化,因此,本实验选择较低温度80 ℃作为提取温度。
2.2 质谱条件的优化
设置蠕动泵转速0.3 r/s;分析模式为高灵敏度;采集模式TRA;75As积分时间0.5 s,同时监测35Cl是否造成干扰,积分时间0.01 s;比较调谐模式No Gas模式和He模式,显示采用No Gas模式时灵敏度高,且无干扰,无需He模式。
2.3 色谱条件的优化
2.3.1 色谱柱的选择 砷形态分析常采用阴离子交换色谱柱Dionex IonPac A19和PRP X-100,且以IonPac A19应用较多,但其分离AsB和AsC相对困难,常无法分离或分析时间过长[11],而PRP X-100则可以较好地解决此问题。
2.3.2 流动相的选择 磷酸盐溶液体系和碳酸盐溶液是砷形态分析常用的流动相,近年多采用碳酸铵溶液,但其对As(V)的洗脱时间长,常需要梯度洗脱[7,12]。由于阴离子色谱柱的柱长、柱体积较大,若柱平衡时间长,梯度洗脱会影响基线稳定性,因此仅在磷酸盐体系范围内选择流动相。参照文献[6,13-14],分别采用50 mmol/L (NH4)2HPO4(pH 7.5),50 mmol/L (NH4)2HPO4(pH 8.0)和20 mmol/L磷酸缓冲溶液 (pH 6.15)进行试验,未能实现6种砷形态的基线分离。
分别选择不同磷酸盐体系(NH4H2PO4,(NH4)2HPO4和NaH2PO4)、改变磷酸盐溶液的浓度(10~50 mmol/L)、溶液pH值(5.0~9.0)等条件,进行流动相A正交试验,正交试验设计见表2。文献报道,少量乙醇对砷元素检测有增敏作用[15],以乙醇为流动相B,VA∶VB=99∶1。3个具有代表性的实验条件对应的色谱图见图1。
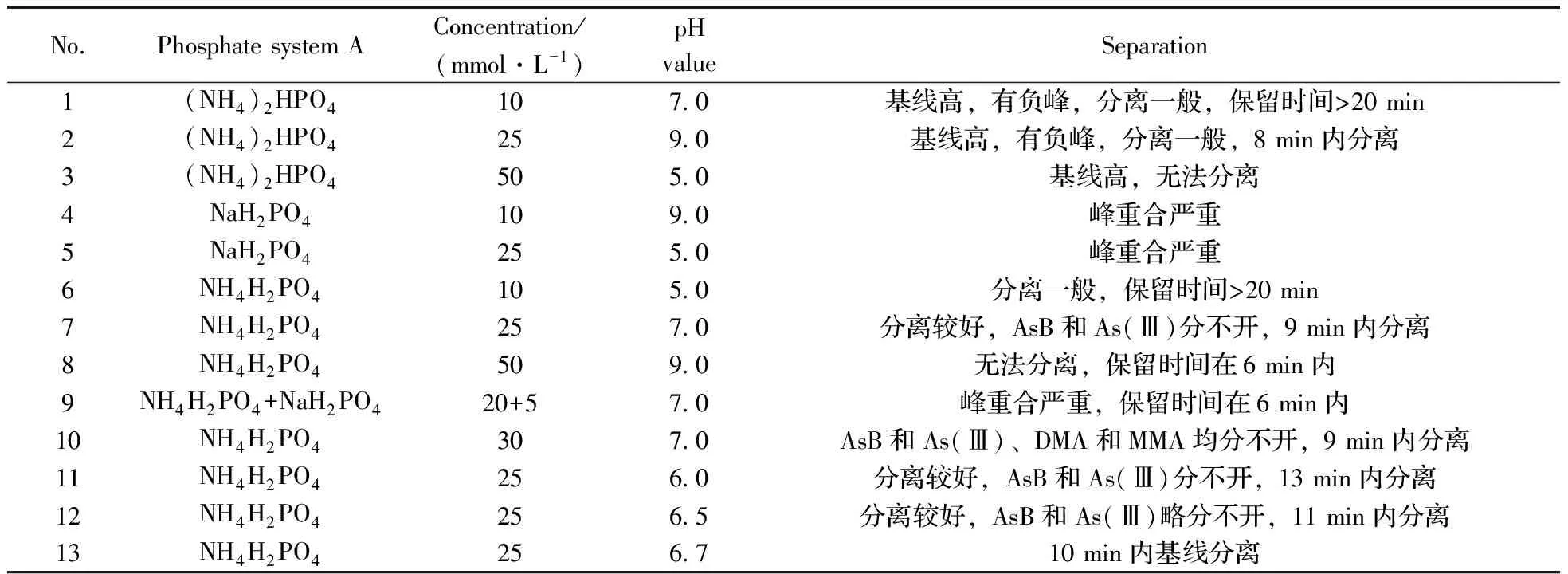
表2 流动相A正交试验

图1 不同流动相A(A∶乙醇=99∶1)洗脱时6种砷形态混合标准溶液的色谱图Fig.1 Chromatograms of six mixed standard arsenic species with different mobile phases A(A∶ethanol=99∶1)a.25 mmol/L (NH4)2HPO4;b.20 mmol/L NH4H2PO4+5 mmol/L NaH2PO4;c.25 mmol/L NH4H2PO4 (pH 6.7)
结果表明,(NH4)2HPO4溶液体系的基线很高,图1a条件下基线CPS计数达到4.8×105,稳定性和灵敏度降低;NaH2PO4可使洗脱速度明显增加,图1b中AsB和AsC几乎同时出峰,无法分离;随着NH4H2PO4溶液浓度的增加,各砷形态的洗脱速度均有不同程度增加,最后出峰的As(V)变化最为明显,NH4H2PO4溶液浓度达到25 mmol/L时可在10 min内洗脱完全,且分离度最好,继续增加NH4H2PO4浓度,则无法实现基线分离;用氨水调节溶液pH值,随着pH值的增加,AsB,AsC和As(Ⅲ)的保留时间延长,而DMA,MMA和As(V)的保留时间缩短,pH 6.7时分离度最好(图1c)。因此选择25 mmol/L NH4H2PO4溶液(pH 6.7)-乙醇(99∶1)作为流动相,6种砷形态可在10 min内实现基线分离,保留时间见表3。
2.4 标准曲线、检出限与精密度
砷形态混合标准溶液的系列浓度分别为0,10.0,20.0,40.0,80.0,100.0 μg/L;总砷标准溶液系列浓度分别为0,1.0,5.0,10.0,50.0,100.0 μg/L,以75As的浓度(X,μg·L-1)为横坐标,75As的CPS计数(Y)为纵坐标建立线性回归方程。以3倍信噪比(S/N=3)计算各砷形态的检出限。以40 μg/L混合标准溶液连续进样6次测定结果的相对标准偏差(RSD)为精密度。结果列于表3。

表3 各砷形态的保留时间、线性方程、相关系数、检出限及相对标准偏差(n=6)
(续表3)

ArsenicspecieRetentiontime/minRegressionequationCorrelationcoefficient(r2)Detectionlimit/(μg·kg-1)RSD/%DMA3 226Y=4194 6X0 99950 450 96MMA3 668Y=4348 8X0 99960 441 3As(V)8 937Y=4507 9X0 99971 02 0
结果显示,6种砷形态的检出限均不大于1.0 μg/kg,精密度均不大于2.0%,各砷形态和总砷的标准曲线线性良好,相关系数为0.999 5~0.999 7,线性范围为1.0~100.0 μg/L。
2.5 加标回收率
向鸡内金和水蛭样品中加入40 μg/L混合标准溶液进行加标回收率实验,平行测定3次,结果列于表4。其平均回收率为94.2%~103.8%,可知采用微波酸提取方法,各砷形态基本保持不变,稳定性好。

表4 鸡内金和水蛭砷形态的加标回收率(n=3)
ND:no detected;a:ventriculi galli mucosa,b:leech
2.6 实际样品的测定结果及提取率
采用本方法分析地龙、水蛭、海螵蛸、桑螵蛸、石决明、鸡内金中的各砷形态含量、提取态砷总量和总砷含量,并计算回收率和提取率(表5),图2为各砷形态的色谱图。

表5 6种中药样品中砷形态和总量的测定结果及提取率
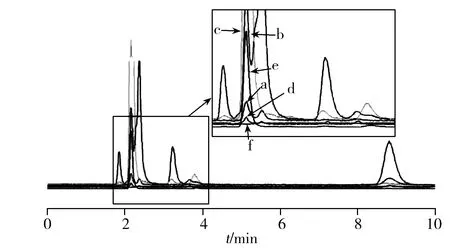
图2 6种中药样品中砷形态的色谱图Fig.2 Chromatograms of arsenic species in six traditional Chinese medicine samplesa.pberetima,b.leech,c.cuttlebone,d.ootheca mantidis,e.shell of abalone,f.ventriculi galli mucosa
由分析结果可以看出,6种动物源性中药的砷形态比例各有不同,石决明和海螵蛸中的无机砷含量均低于砷总量的1%,砷主要以有机形态存在,这与海洋生物中有机砷比例很高的报道一致[16];从表5数据可计算得到,其他4种样品中的无机砷含量为50.6%~76.6%,无机砷比例较高,说明在陆地生物体内砷形态转化比例有限。而动物源性中药绝大部分仍来源于陆地生物,使用时需注意砷毒性。
方法提取率即提取态砷与总砷质量浓度之比,本方法的提取效率为95.5%~102.3%,提取效果良好。除水蛭外,5种样品中各形态砷的含量之和与其经消解测得的提取态砷含量基本相符,说明可提取砷各形态均被检出。水蛭中AsB和As(Ⅲ)未能实现基线分离,从峰形来看,两峰之间应含有另一种砷形态,可能是一种砷糖。因没有合适的标准品,为验证这个推论,按实验方法处理藻类样品,同样检出1个含量极高的未知峰,保留时间与水蛭可能存在峰的保留时间相同。文献报道藻类样品中砷主要以砷糖形态存在[16],因此水蛭中未能检出的砷形态可能为藻类中所含砷糖。该砷形态的存在干扰了水蛭中AsB和As(Ⅲ)的基线分离,但从加标回收率来看,并未影响定量结果,不影响该方法的应用范围。可以尝试进一步摸索实验方法,实现砷糖与这6种砷形态的色谱分离。
3 结 论
本文建立了微波酸提取/LC-ICP-MS测定动物源性中药中6种砷形态的分析方法,比较并优化了提取条件和流动相,在等度洗脱条件下,10 min内可实现基线分离,并进行了方法学考证。对实际样品的分析可知,微波酸提取方法简便、快速,整个前处理过程用时仅需1 h,提取率高,各砷形态稳定,重现性好,可应用于多种动物源性中药及含有相关成分的中成药中砷形态的分析,为药材质量监督及批量、快速检测提供方法参考。动物源性中药中的砷元素主要以这6种形态存在,极少数水生物中药材可能含有砷糖成分,其具体结构有待于进一步考证。
[1] Li C Y,Zhang Y Y.J.Instrum.Anal.(李春盈,张玉英.分析测试学报),2016,35(12):1634-1638.
[2] Chinese Pharmacopoeia Commission.Chinese Pharmacopoeia.2015 ed,PartⅠ.Beijing:China Medical Science Press(国家药典委员会.中国药典.一部,2015版.北京:中国医药科技出版社),2015:83,122,294.
[3] Hsieh Y J,Jiang S J.J.Agric.FoodChem.,2012,60:2083-2089.
[4] Zhang M,Zhang R X,Wu P,Guan Q S.J.Anal.Sci.(张萌,张瑞雪,吴攀,官庆松.分析科学学报),2015,31(4):484-488.
[5] Zu W C,Wang Y,Liu C,Chang J L,Li M Z.Chin.J.Anal.Lab.(祖文川,汪雨,刘聪,常久乐,李明章.分析试验室),2016,35(1):82-85.
[6] Chen S Z,Liu L P,Du Z X,Ma H.J.Chin.MassSpectrom.Soc.(陈绍占,刘丽萍,杜振霞,马辉.质谱学报),2015,36(1):33-39.
[7] Lu Y N,Chen J W,Zhang L T,Wei J H,Lin W.J.Anal.Sci.(陆奕娜,陈建伟,张林田,魏建华,林文.分析科学学报),2016,32(1):141-144.
[8] Chinese Pharmacopoeia Commission.Chinese Pharmacopoeia.2015 ed,Part Ⅳ.Beijing:China Medical Science Press(国家药典委员会.中国药典.四部,2015版.北京:中国医药科技出版社),2015:207.
[9] Ni Z L,Tang F B,Yu Q,Mo R H.Chin.J.Anal.Lab.(倪张林,汤富彬,喻晴,莫润宏.分析试验室),2014,33(12):1392-1396.
[10] Wang C Y,Li L X,Xie T T,Lin J F,Chu N Q.J.Instrum.Anal.(王成云,李丽霞,谢堂堂,林君峰,褚乃清.分析测试学报),2016,35(12):1554-1562.
[11] Yang L J,Hu Q R,Guo W,Liu Y M,Song X H,Zhang P C.Chin.J.Chromatogr.(杨丽君,胡巧茹,郭伟,刘玉敏,宋晓华,张鹏程.色谱),2011,29(5):394-398.
[12] Cao X,Li J X,Yu J J,Yu Z H,Yang H H,Wang X R.J.Instrum.Anal.(曹煊,李景喜,余晶晶,于振花,杨黄浩,王小如.分析测试学报),2009,28(3):257-261.
[13] Li L M,Xia J,Wang X M,Wang M B,Wang K,Ji S.Chin.Tradit.Pat.Med.(李丽敏,夏晶,王欣美,王枚博,王珂,季申.中成药),2012,34(11):2118-2123.
[14] Wang L M,Wei C Y.Environ.Chem.(王玲梅,韦朝阳.环境化学),2010,29(4):729-733.
[15] Chen G,Lin L,Chen Y H.Environ.Chem.(陈光,林立,陈玉红.环境化学),2009,28(4):608-611.
[16] Lü C,Liu L P,Dong H R,Li X W.J.Instrum.Anal.(吕超,刘丽萍,董慧茹,李筱薇.分析测试学报),2010,29(5):465-468.
Determination of Six Arsenic Species in Animal Derived Traditional Chinese Medicines by Liquid Chromatography Combined with Inductively Coupled Plasma Mass Spectrometry Assisted by Microwave Acid Extraction
CAO Can*,LIU Cheng-yan,WANG Zhi-jia,WU Zhi-gang,CUI Chang,WU Dong-dong
(Liaoning Analysis Science Academe,Shenyang 110015,China)
A method was established for the determination of six arsenic species including arsenite(As(Ⅲ)),arsenate(As(V)),monomethylarsine (MMA),dimethyarsine (DMA),arsenobetaine(AsB) and arsenocholine(AsC),in animal derived traditional Chinese medicines by liquid chromatography coupled with inductively coupled plasma mass spectrometry(LC-ICP-MS) assisted by microwave acid extration.After microwave extraction with 1% HNO3solution within 10 min and centrifugation,the sample was purified with solid phase extraction SPE C18and 0.45 μm hybrid filter membrane,and then eluted with 25 mmol/L NH4H2PO4(pH 6.7) -ethanol(99∶1,by volume) as mobile phase by isocratic elution.The arsenic species could reach a baseline separation within 10 min.The results showed that the linear ranges for six arsenic species were in the range of 1.0-100.0 μg/L with correlative coefficients of 0.999 5-0.999 7.The limits of detection (LOD) for six anions were in the range of 0.24-1.0 μg/kg with relative standard derivations(RSD) of 0.96%-2.0%.The method was used in the determination of arsenic speciation in six samples involved pberetima,leech,cuttlebon,ootheca mantidis,shell of abalone and ventriculi galli mucosa.The spiked recoveries were between 94.2% and 103.8% and the extraction efficiencies were in the range of 95.5%-102.8%,which are better than those obtained by heat extraction.With the advantages of quickness,accuracy and good reproducibility,the method was suitable for the arsenic species detection and quality monitoring of animal derived traditional Chinese medicine and similar samples.
microwave acid extration;liquid chromatography coupled with inductively coupled plasma mass spectrometry (LC-ICP-MS);animal derived traditional Chinese medicine;arsenic species analysis
2017-02-10;
2017-03-10
辽宁省自然科学基金项目( 20170540481);辽宁省科学事业公益研究基金项目( 2016002003)
10.3969/j.issn.1004-4957.2017.06.009
O657.63;O613.63
A
1004-4957(2017)06-0756-06
*通讯作者:曹 璨,硕士,副研究员,研究方向:光谱、质谱分析,Tel:024-24210497,E-mail:sining1998@126.com
