阿卡波糖中杂质的分离富集方法研究
2017-06-28梁现蕊张会晨何小嫒苏为科
梁现蕊,张会晨,何小嫒,吴 晖,苏为科
(1.浙江工业大学 药学院,浙江 杭州 310014;2.杭州中美华东制药股份有限公司,浙江 杭州 310011)
阿卡波糖中杂质的分离富集方法研究
梁现蕊1,2,张会晨1,何小嫒1,吴 晖2,苏为科1
(1.浙江工业大学 药学院,浙江 杭州 310014;2.杭州中美华东制药股份有限公司,浙江 杭州 310011)
采用不同的酸、碱条件对阿卡波糖进行降解反应,可得到其杂质A和D,不同酸碱浓度、温度和降解时间等条件对两种杂质的生成量有较大的影响.结果显示在30 ℃和0.5 moL/L的NaOH溶液中降解3 h杂质A组分产量较高;在25 ℃和12 moL/L的HCl溶液中降解1 h杂质D组分产量较高.降解实验得到的粗品杂质A和D经制备液相色谱进一步分离富集,分别得到高纯度的两种杂质,并采用高分辨质谱对其进行了结构确认.实验对于提高阿卡波糖产品质量标准具有重要意义.
阿卡波糖;杂质A;杂质D;分离富集
阿卡波糖(acarbose)是α-葡萄糖苷酶抑制剂类降糖药,是治疗II型糖尿病(非胰岛素依赖型糖尿病)的首选药[1].II型糖尿病患者自身可以分泌胰岛素,但胰岛素分泌量不足或者外周组织胰岛素敏感性降低,使患者血糖变化较大,II型糖尿病占患者的90%,因此II型糖尿病的治疗药物是降糖药研究的重点[2].阿卡波糖跟寡糖的结构类似,能竞争性地与α-葡萄糖苷酶结合,减少寡糖的消化吸收,从而达到降低餐后血糖水平的目的[3].
阿卡波糖的工业生产采用微生物发酵法合成,是由游动放线菌合成的假四糖类次级代谢物,阿卡波糖产品主要从其发酵液中分离制得.然而,发酵过程中除了生成阿卡波糖外,还产生了一些阿卡波糖结构类似物,这些类似物成为阿卡波糖中杂质的主要来源[4-5].药物中的杂质是影响药物纯度的主要因素,杂质的存在直接影响到药物的安全性、有效性和稳定性,将其控制在一个安全、合理的限度范围之内,将直接关系到上市药品的质量及安全性[6].因此,对阿卡波糖中的杂质进行研究,将对提升阿卡波糖产品质量标准具有重要意义.在杂质的分离分析方法中,高效液相色谱-质谱联用(HPLC-MS)技术集液相色谱的高分离能力与质谱的强结构解析能力于一体,已成为药物杂质分析的有力工具[7-9].目前,已有一些利用离子交换树脂分离富集阿卡波糖的报道[10-11],但是鲜有关于杂质A和杂质D分离富集方法的报道,为了获得高纯度的杂质A和D,本实验开发了杂质的富集方法,结合制备液相色谱进一步对杂质产物进行了分离制备[12-14],获得了高纯度的单一杂质产品,为后续阿卡波糖杂质的药理、毒理学研究提供材料,也为准确控制杂质含量提供了依据.
1 实验部分
1.1 材料与试剂
阿卡波糖由杭州中美华东制药股份有限公司提供.乙腈(HPLC级,Merck),实验用其他试剂均为分析纯,购自天津市永大化学试剂有限公司;实验室用超纯水(18.2 MΩ)由Barnstead TII超纯水系统制得.
1.2 仪器及设备
Waters Delta 600半制备液相色谱仪(Waters,美国);Agilent 1100液相色谱-Finnigan Advantage LCQ质谱联用仪(Thermo Scientific,美国);Bruker micrOTOF-Q II串联四极杆-飞行时间质谱仪(Bruker,美国);Barnstead TII超纯水系统(Thermo Scientific,美国).
1.3 实验方法
1.3.1 阿卡波糖液相色谱检测条件
取阿卡波糖样品用纯水溶解,配置成质量浓度为2 mg/mL的溶液,参照欧洲药典,以磷酸缓冲盐和乙腈作为流动相,进行液相色谱检测[15];色谱柱为Welch Ultimate XB-NH2(4.6 mm×250 mm,5 μm),检测波长210 nm,流动相组成为V(0.6 g/L的磷酸二氢钾与0.35 g/L的磷酸氢二钠水溶液)∶V(乙腈)=30∶70,进样量5 μL,柱温30 ℃,流速1.0 mL/min.
1.3.2 液质联用分析条件
高效液相色谱部分为Aglient 1100,液相条件同1.3.1,但将其流动相中的磷酸盐缓冲溶液换成纯水,以免不挥发盐堵塞质谱的毛细管.喷雾电压4 kV,毛细管温度300 ℃,毛细管电压24 V,扫描范围m/z为50~1 000,正负离子模式下检测.
1.3.3 串联四级杆飞行时间质谱分析条件
高分辨质谱(HRMS)数据由OTOF-MS检测得到,质量数精确到小数点后四位并能模拟出分子式.电喷雾离子源(ESI),正离子模式下扫描,毛细管电压4 500 V,氮气(N2)作为干燥气和雾化气,干燥气温度180 ℃,雾化器压力40 kPa,干燥气流速4.0 L/min,用浓度为0.01 mol/L的三氟乙酸钠溶液校正质量范围.
1.3.4 阿卡波糖的酸降解液制备
将阿卡波糖样品加入到12 mol/L的盐酸溶液中,配成质量浓度为40 mg/mL的溶液,于恒温培养箱中控制温度为25 ℃,静置1 h,用氢氧化钠溶液中和至中性.
1.3.5 阿卡波糖的碱降解液制备
将阿卡波糖样品加入到0.5 mol/L的氢氧化钠溶液中,配成质量浓度为40 mg/mL的溶液,于恒温培养箱中控制温度为30 ℃,静置3 h,用盐酸溶液调节pH值至中性.
1.3.6 制备液相色谱制备杂质A和D
通过酸碱降解实验得到高杂产物,使用制备液相分离富集杂质.所用制备型色谱柱为Welch Ultimate XB-NH2(210 mm×250 mm,5 μm),流速为21 mL/min,检测波长为210 nm,柱温为室温,进样质量浓度40 mg/mL,进样量500 μL,流动相为V(水)∶V(乙腈)=30∶70.
2 结果分析与讨论
实验首先采用HPLC-MS技术对阿卡波糖降解液中的成分进行了检测,以便下一步对阿卡波糖及其杂质进行定位分析.
2.1 HPLC-MS用于阿卡波糖降解液成分分析
2.1.1 酸降解结果的分析
将酸降解液进行液质联用分析,HPLC色谱图和基峰离子流图如图1,2所示.结果显示:在保留时间27 min处出现646.2的离子峰,保留时间、离子峰结果均与阿卡波糖一致,因此判断为阿卡波糖.在保留时间16.3 min处出现484.2的离子峰,根据相关文献报道[2],杂质D相对阿卡波糖的保留值是0.6,初步判断保留时间16.3 min处是杂质D的[M+H]+峰.
对保留时间16.3 min处484.2的离子峰进行质谱二级裂解,通过调节质谱的锥孔电压,得到了m/z=304.1碎片离子峰,说明该物质失去一个质量分数为180的碎片后生成m/z=304.1的离子,即杂质D脱去一个环的过程.对m/z=304.1的离子进行三级裂解,得到m/z=146.0的碎片,推测其裂解过程为
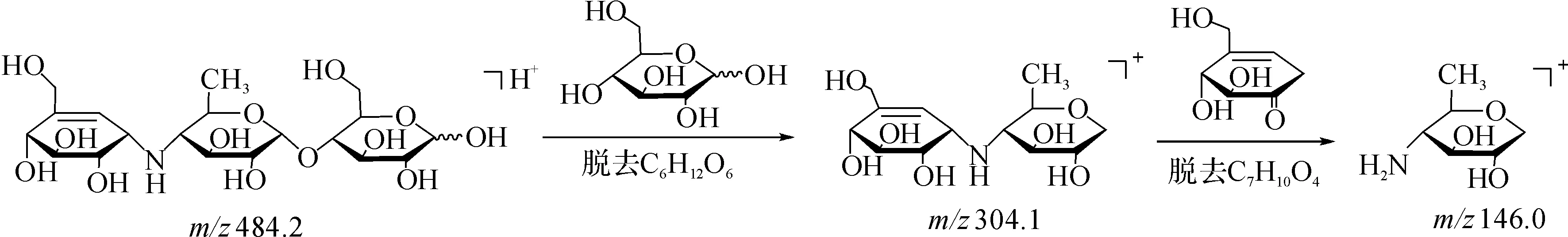

图1 酸降液的液相色谱图Fig.1 HPLC chromatography after acid degradation
图2 液质的基峰离子流图Fig.2 Base peak ion chromatogram after acid degradation
2.1.2 碱降解结果的分析
将碱降解液进行液质联用分析,HPLC色谱图和基峰离子流图见图3,4,结果显示:保留时间在25.2 min和27.0 min处均出现646.2的离子峰.根据相关文献报道[2],阿卡波糖和杂质A的分子量相同,而杂质A相对阿卡波糖的保留值是0.9.因此,根据分子量及相对保留时间推测25.2 min处是杂质A的[M+H]+峰,27.0 min是阿卡波糖的[M+H]+峰.
图3 碱降液的液相色谱图Fig.3 HPLC chromatography after alkali degradation

图4 碱降液的基峰离子流图Fig.4 Base peak ion chromatogram after alkali degradation
对646.2的离子峰进行质谱二级裂解,保留时间在25.2 min和27.0 min的峰裂解后,均得到了m/z=304.1的碎片离子峰.对m/z=304.1的峰进行三级裂解,得到m/z=146.0的碎片离子峰,根据阿卡波糖和杂质A的结构可知:它们均有四个糖环,具有相类似的裂解途径即阿卡波糖和杂质A脱环的过程,推测杂质A裂解过程为
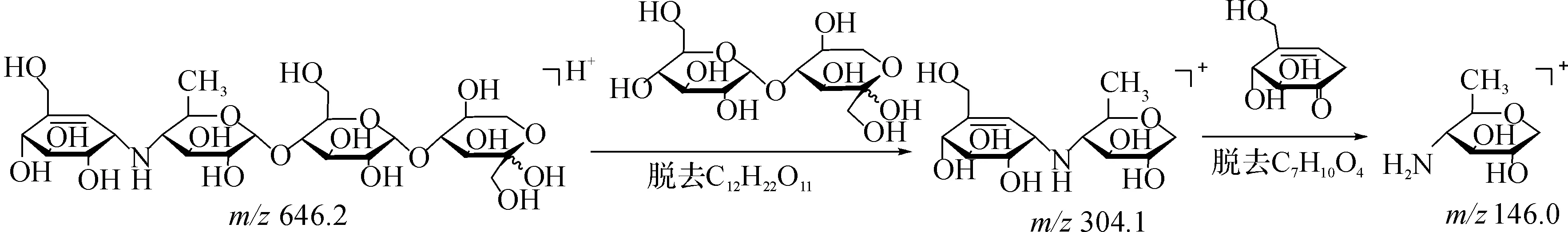
在上述确定杂质A和D的相对保留时间的基础上,进一步考察富集杂质的相关条件.
2.2 不同降解条件对杂质D生成量的影响
2.2.1 酸浓度对降解实验的影响
实验分别取浓度为12,10,8 mol/L的盐酸溶液,对阿卡波糖进行降解实验,恒温培养箱控制温度为25 ℃,1 h后用氢氧化钠溶液中和至中性,HPLC检测杂质峰的变化.结果显示,随着盐酸浓度的降低,杂质D的质量分数也在降低,阿卡波糖不能彻底降解,所以12 mol/L盐酸为较佳酸降解浓度.
2.2.2 温度对酸降解实验的影响
实验取阿卡波糖样品5组,加入12 mol/L的盐酸溶液,配置成质量浓度为40 mg/mL的溶液,在恒温培养箱中分别以15,20,25,30,35 ℃的温度静置1 h,HPLC检测结果显示,25 ℃时杂质D质量分数最高,温度过低导致阿卡波糖不能很好的降解,温度过高会使阿卡波糖过度分解生成其他杂质,故酸降解反应的较佳温度为25 ℃.
2.2.3 反应时间对酸降解实验的影响
进一步考察酸降解时间对杂质D质量分数的影响,采用12 mol/L盐酸溶液为降解液,在25 ℃恒温培养箱中静置反应8 h;定时取样,氢氧化钠溶液中和后进行液相色谱分析,结果如表1所示.

表1 阿卡波糖酸降解实验结果表1)
注:1) 峰面积由面积归一化法计算得到.
实验结果表明:在相同酸浓度和反应温度下,随着反应时间的增加,杂质D质量分数是在不断降低的,阿卡波糖的量也在降低,强酸性条件使杂质D和阿卡波糖都发生了降解,所以得到杂质D粗品的较佳降解条件降解温度为25 ℃、降解时间为1 h、降解液为12 mol/L的盐酸溶液,杂质D的质量分数能达到37.3%.
2.3 不同降解条件对杂质A生成量的影响
2.3.1 碱浓度对降解实验的影响
实验分别取0.5,1.0,1.5 mol/L的氢氧化钠溶液,对阿卡波糖进行降解实验,2 h后用盐酸溶液中和至中性,HPLC检测杂质峰的变化.结果显示,随着氢氧化钠浓度的升高,杂质A的质量分数在降低,碱浓度为0.5 mol/L更有利于阿卡波糖降解转化得到杂质A,所以0.5 mol/L氢氧化钠为较佳碱降解浓度.
2.3.2 温度对碱降解实验的影响
实验取阿卡波糖样品5组,加入0.5 mol/L的氢氧化钠溶液,在恒温培养箱中分别以15,20,25,30,35 ℃的温度静置2 h.HPLC检测结果显示,30 ℃时杂质A质量分数最高,温度过低导致阿卡波糖不能很好的降解,温度过高会使阿卡波糖过度分解生成其他杂质,故降解反应的较佳温度为30 ℃.
2.3.3 反应时间对碱降解实验的影响
进一步考察碱降解时间对杂质A生成量的影响,采用0.5 mol/L氢氧化钠溶液为降解液,在30 ℃恒温培养箱中静置反应8 h;定时取样,用盐酸溶液中和至中性,进行液相色谱分析,结果如表2所示.
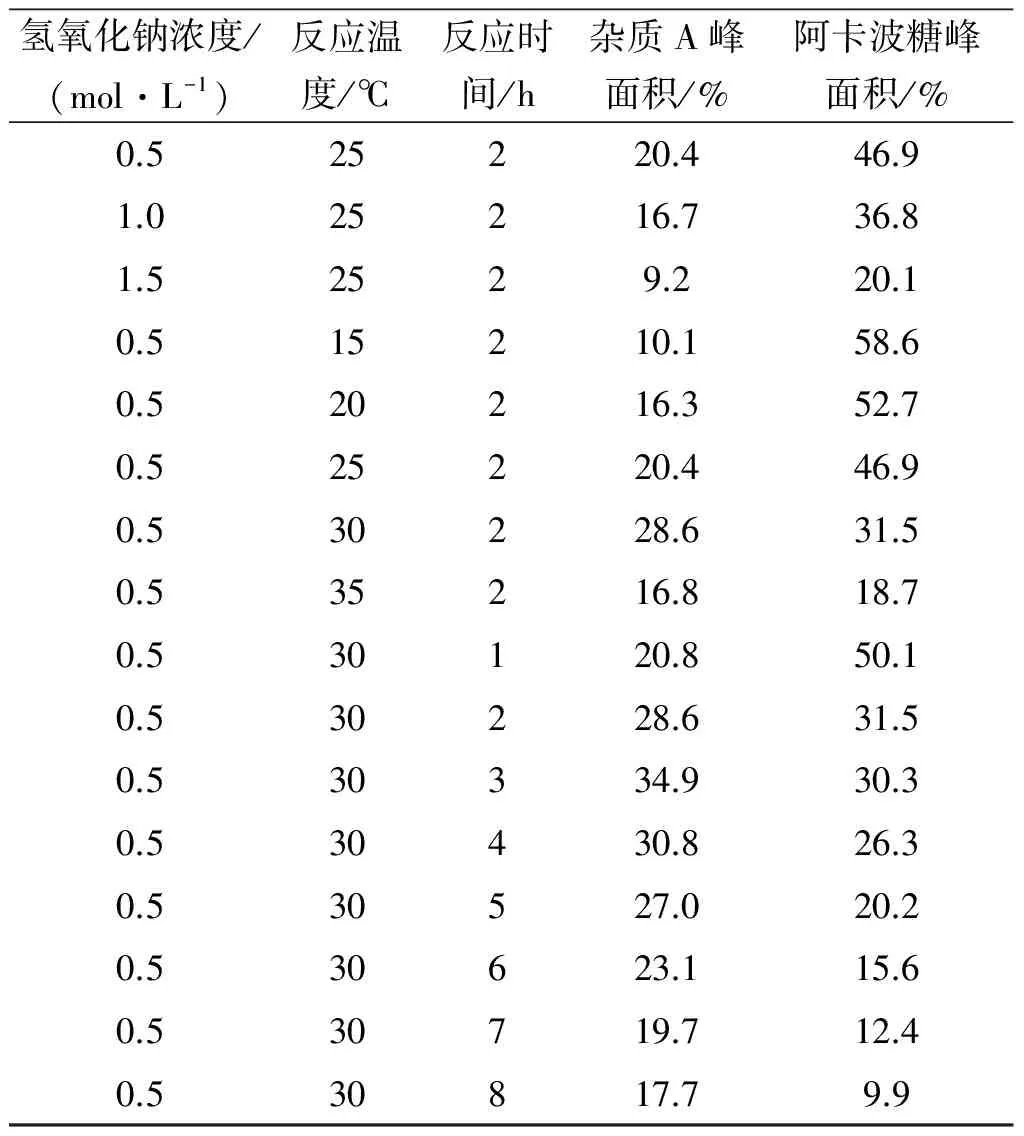
表2 阿卡波糖碱降解实验结果表1)
注:1) 峰面积由面积归一化法计算得到.
如表2所示,其他条件不改变,反应时间3 h得到杂质A的质量分数最高,达到了34.9%.综合以上实验结果,优化后的阿卡波糖碱降解得到杂质A的实验条件为降解温度30 ℃,降解时间3 h,降解溶液0.5 mol/L的氢氧化钠.
2.4 杂质的分离纯化与结构表征
用制备液相对酸降解后质量分数为37.3%的杂质D降解液进行分离纯化,结果得到质量分数为98.7%杂质D;对碱降解后质量分数为34.9%的杂质A降解液进行分离纯化,得到质量分数为98.9%杂质A;杂质D和A经高分辨质谱进一步表征,结果如下:
1) 杂质D为白色粉末,在其HRESI-MS正离子质谱图中,在m/z=484.203 7处具有较强的离子峰,为样品的[M+H]+,该离子的准确分子式为C19H34NO13,说明样品的准确分子式为C19H33NO13,与杂质D的分子量和分子式一致.
2) 杂质A为白色粉末,在其HRESI-MS正离子质谱图中,在m/z=646.256 5处具有较强的离子峰,为样品的[M+H]+,该离子的准确分子式为C25H44NO18,说明样品的准确分子式为C25H43NO18,与杂质A的分子量和分子式一致.杂质D和A的结构式分别为
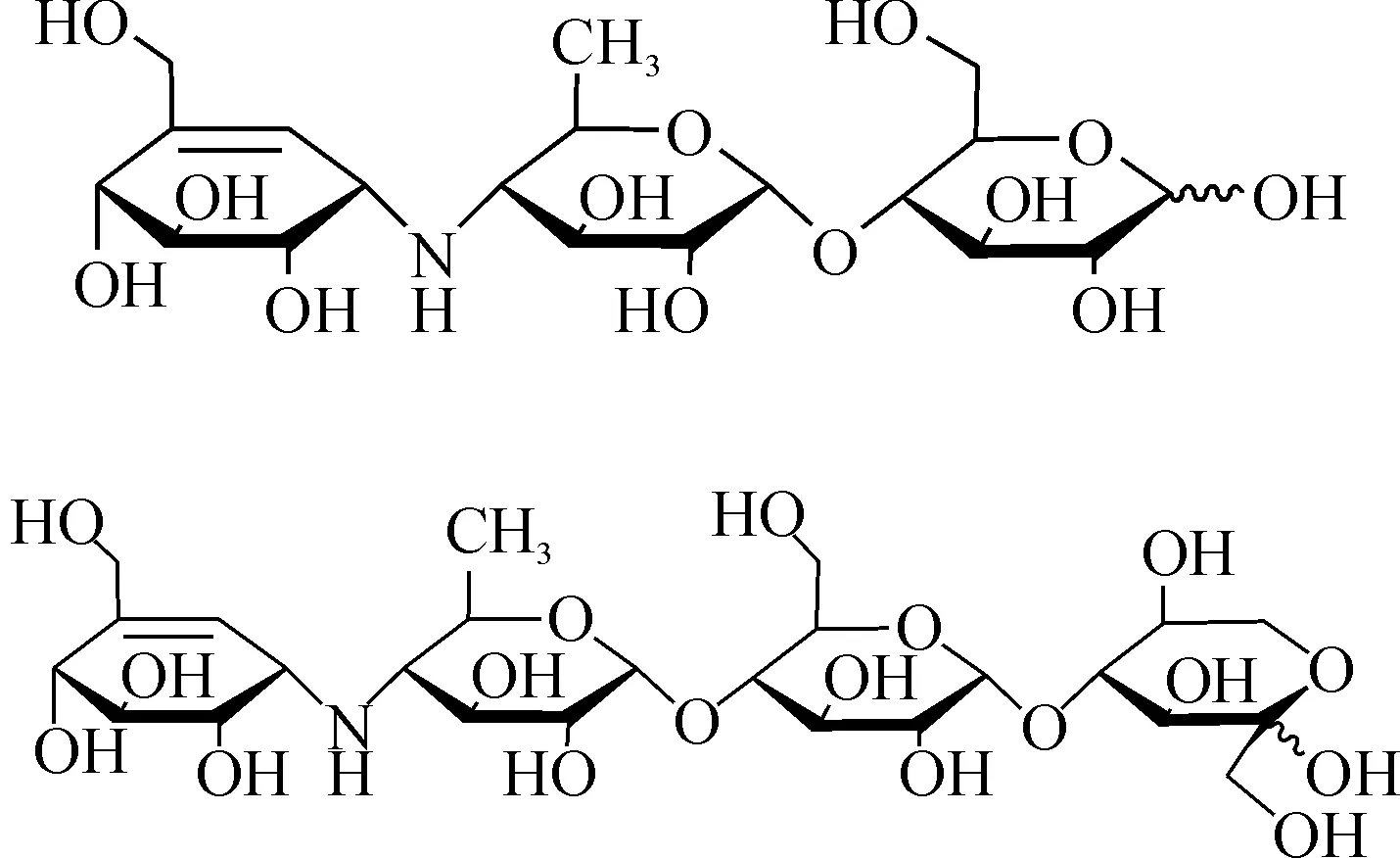
上述高分辨质谱结果并结合液质联用质谱裂解碎片分析结果证明,分离得到的两个化合物分别是杂质D和杂质A,与欧洲药典报道的杂质D和杂质A结构一致.
3 结 论
采用HPLC-MS方法,能有效地鉴别出杂质的保留时间及分子量,可对杂质进行定性分析.实验通过对不同酸碱浓度、温度和降解时间进行筛选,研究了降解富集杂质A和D的较佳条件,并利用半制备液相色谱技术进一步分离纯化得到两个杂质,该分离富集方法可获得较高纯度的杂质单体,且经结构鉴定证实其为预期的杂质.实验所获得的杂质A和D可作为标准物质用于后续的杂质其他方面研究.另外,本研究可为药物杂质分析方法提供一条可借鉴的研究思路,同时为提升阿卡波糖质量标准提供了有力的帮助.
[1] 孙楠,周雪枫,胡宝祥,等.阿卡波糖的单扫描示波极谱法测定研究[J].浙江工业大学学报,2005,33(6):614-617.
[2] WANG Yajun, ZHENG Yuguo, XUE Yaping, et al. Analysis and determination of anti-diabetes drug acarbose and its structural analogs[J].Current pharmaceutical analysis,2011,7(1):12-20.
[3] RODRIGUEZ J F, LUCAS A D, CARMONA M, et al. Application of ion exchange to purify acarbose from fermentation broths[J].Biochemical engineering journal,2008,40:130-137.
[4] RAUT B B, KOLTE B L, DEO A A, et al. Quantification of acarbose in human plasma by liquid chromatography-electrospray tandem mass spectrometry[J]. Journal of liquid chromatography & related technologies,2004,27(11):1759-1768.
[5] 黄剑川,王超儿,王亚军.阿卡波糖结构类似物组分A形成机制研究及其制备[J].发酵科技通讯,2014,43(4):1-6.
[6] 祝清芬,李涛,国明,等.药物杂质的毒理学评价要求及进展[J].中国新药杂志,2010,19(24):2271-2276.
[7] LIANG Xianrui, XU Qiao. Separation and identification of phenolic compounds in Bidens pliosa L. by ultra-performance liquid chromatography coupled with quadrupole time-of-flight mass spectrometry[J].Journal of separation science,2016,39(10):1853-1862.
[8] NOVAK P, TEPES P, CINDRIC M, et al. Combined use of liquid chromatography-nuclear magnetic resonance spectroscopy and liquid chromatography-mass spectrometry for the characterization of an acarbose degradation product[J].Journal of chromatography A,2004,1033:299-303.
[9] NOVAK P, CINDRIC M, TEPES P, et al. Identification of impurities in acarbose by using an integrated liquid chromatography-unclear magnetic resonance and liquid chromatography-mass spectrometry approach[J].Journal of separation science,2005,28:1442-1447.
[10] WANG Yajun, YU Lei, ZHENG Yuguo, et al. Acarbose isolation with gel type strong acid cation exchange resin: equilibrium, kinetic and thermodynamic studies[J].Separation science and engineering,2013,21(10):1106-1113.
[11] 王亚军,董方智,于蕾,等.阳离子交换树脂SAC 001×7对阿卡波糖的吸附性能研究[J].高校化学工程学报,2012,26(3)493-498.
[12] 张翠萍,李行诺,陈鸳谊.反相高效制备液相色谱法制备洋川芎内酯H和I[J].浙江工业大学学报,2011,39(4):386-389.
[13] 刘小琳,宋丽明,雷勇胜,等.制备型高效液相色谱法在药物杂质研究中的应用[J].药物评价研究,2012,35(5):233-236.
[14] 梁现蕊,赵萃.韩信草化学成分分离与结构鉴定[J].浙江工业大学学报,2016,44(1):88-91.
[15] European Pharmacopoeia Commission.European pharmacopoeia[M]. Strasbourg: Council of Europe,2011:1302-1303.
(责任编辑:刘 岩)
Isolation and preparation methods of acarbose impurities
LIANG Xianrui1,2, ZHANG Huichen1, HE Xiaoai1, WU Hui2, SU Weike1
(1.College of Pharmaceutical Sciences, Zhejiang University of Technology, Hangzhou 310014, China; 2.Hangzhou Zhongmei Huadong Pharmaceutical Co., Ltd., Hangzhou 310011, China)
The degradation of acarbose in acid and alkaline solutions can produce impurity A and D. Concentrations of acid and alkali, temperature and reaction time have great influences on the production of two impurities. The results showed that the degradation time of 3 h at 30 ℃ in 0.5 NaOH moL/L solution could produce higher production of the impurity A. Higher production of the impurity D was obtained with the degradation time of 1 h at 25 ℃ in 12 moL/L HCl solution. The impurities A and D were further isolated and purified by preparative high-performance liquid chromatography, and their structures were confirmed by high resolution mass spectra. The experiment has an important significance for improving the acarbose’s quality standard.
acarbose; impurity A; impurity D; isolation and preparation
2016-11-04
浙江省博士后择优资助科研项目(BSH1502011)
梁现蕊(1975—),女,山东临沂人,副教授,博士,研究方向为药物结构鉴定与杂质的分离分析,E-mail:liangxrvicky@zjut.edu.cn.
R917
A
1006-4303(2017)03-0289-05
