烯烃配位聚合催化剂和聚合反应
2017-06-27胡友良
胡友良
(中国科学院 化学研究所 工程塑料重点实验室,北京100190)
特约述评
烯烃配位聚合催化剂和聚合反应
胡友良
(中国科学院 化学研究所 工程塑料重点实验室,北京100190)
对烯烃配位聚合发展状况进行了简要评述,主要内容包括烯烃配位聚合催化剂和聚合反应,并对过去在此领域的相关研究工作进行了简单介绍。涉及的催化剂有Ziegler-Natta催化剂、茂金属催化剂和非茂金属催化剂;介绍的聚合反应有功能化聚烯烃的制备,原位共聚合反应制备线型低密度聚乙烯,链穿梭聚合反应制备烯烃嵌段共聚物,配位链转移聚合反应制备端基功能化聚烯烃。最后,对聚烯烃的研发提出几点建议。
聚烯烃;配位聚合;Ziegler-Natta催化剂;茂金属催化剂;非茂金属催化剂
自从Ziegler在1953年发现配位聚合催化剂以来,烯烃的配位聚合技术就一直保持着高速的发展,从催化剂、聚合方法到聚合工艺,大约每隔十年左右,在技术上就有一次大的突破。由于聚烯烃材料具有巨大的商业应用价值,烯烃的配位聚合吸引了不同学科的科学家为之奋斗,其中包括有机金属化学家、聚合物化学家和物理、分析化学家等。他们在促进烯烃配位聚合发展的同时,也带动了这些学科本身的长足进步。
本文拟结合本人五十年来的研究工作实践,就烯烃配位聚合发展状况做简要评述,主要包括烯烃配位聚合催化剂和聚合反应,也涉及到新的聚合方法及聚烯烃新产品等。
1 烯烃配位聚合催化剂
催化剂是烯烃配位聚合的核心技术。从结构性能的关系来说,催化剂结构决定了聚烯烃的结构(包括相对分子质量、相对分子质量分布、共聚单体含量和分布以及聚合物的规整性等),聚烯烃的微结构又决定了它的宏观性能,从而影响了它的加工性能和应用领域。多年来,烯烃配位聚合催化剂的发展促进了聚烯烃工业的进步。
1.1 Ziegler-Natta催化剂
到目前为止,Ziegler-Natta催化剂还一直是聚烯烃工业使用的主要催化剂,约占95%。Ziegler-Natta催化剂长盛不衰的原因得益于它成熟的工业实践和不断革新。以丙烯聚合用Ziegler-Natta催化剂为例,论述它的制备方法和内外给电子体的发展。
1.1.1 用化学反应法制备高效催化剂载体
研究表明,将Ti化合物负载在载体上可以大幅提高催化剂的活性,主要原因是Ti化合物可以高效分散在载体表面上,增加了催化剂活性中心的浓度;同时,由于Mg的电负性比Ti低,通过推电子效应,使催化活性中心Ti的电子云密度增大,从而降低烯烃分子在活性中心Ti上链增长的活化能,增大链增长速率常数的值。因此,制备合适的载体是提高催化剂活性的关键。本课题组曾用物理研磨的方法制备MgCl2载体,虽然催化剂活性和聚合物等规组分有一定的提高,但还是不能满足工业进步对催化剂的要求。而通过化学反应法制备MgCl2载体,本课题组得到了活性和产物等规组分俱佳的高效聚丙烯催化剂。该方法的基础是MgCl2能与许多种类的Lewis碱(如醇、酯和醚类等化合物)形成可溶于烷烃溶剂的复合物。所谓用化学反应法活化催化剂载体就是先把MgCl2制成这种复合物溶液,再与TiCl4反应,使MgCl2从溶剂中重新析出。可通过控制MgCl2再析出的条件,得到高比表面积和优良结晶形态的高活性载体。本课题组最近系统地综述了Ziegler-Natta催化剂的载体技术研究进展[1]。
1.1.2 化学法制备高效催化剂载体的反应机理
在研发高效催化剂并使之工业化的过程中,本课题组对化学法制备高效催化剂载体的反应机理进行了深入的研究,并通过对制得的催化剂进行结构分析和活性测定,揭示了结构与性能之间的关系[2]。简单来说,载体的制备和活化可以归纳为以下几个反应式:
第一步:固体MgCl2生成复合物溶液
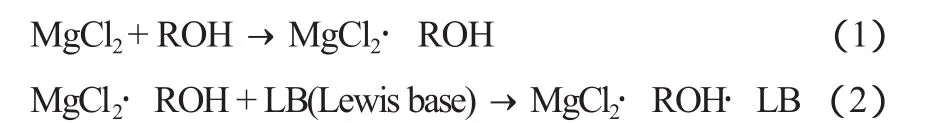
第二步:复合物溶液与TiCl4反应析出MgCl2晶体

从上述反应式可看到,在形成可溶性复合物时加入Lewis碱十分重要。如果没有Lewis碱,析出的晶体[MgCl2(s)]结构为规整的立方密堆积(ccp)和六方密堆积(hcp)形式(反应式5和6);加入了Lewis碱后,就得到MgCl2·LB(反应式4),析出的晶体[MgCl2·LB(s)]的结构是带有旋转无序(rd)的缺陷形式(反应式7)。后者是高效催化剂的最好载体。当前,在工业上使用的MgCl2载体聚丙烯高效催化剂,不管具体的制备方法如何(化学结晶法或球形载体法),用化学法制备载体的反应机理均类似。
1.1.3 纳米球形MgCl2载体
近年来,为了扩展聚烯烃材料的应用领域,制备小尺寸的聚丙烯颗粒越来越受到关注,其中,首要的问题是制备小尺寸的载体。本课题组采用纳米尺寸的球形MgCl2作载体,制备的催化剂具有高活性,能满足工业化生产的要求[3]。
1.1.4 Ziegler-Natta催化体系中的内给电子体
在高效催化剂制备中,加入邻苯二甲酸二酯类化合物作为催化剂的内给电子体确实是一大进步[4-5]。由此,工业上产生了高活性、高等规的聚丙烯催化剂。直至三十年后的今天,大多数工业用催化剂还都使用邻苯二甲酸二酯类化合物作内给电子体,用于生产聚丙烯均聚物和抗冲共聚物等系列产品。
近年来,Ziegler-Natta催化剂的制备工艺改进不多,但内给电子体的创新不少,在此,首先要提及的是新型二醚类化合物[6]。二醚类催化剂的活性更高,大约是二酯类催化剂的两倍以上,而聚合物的立构规整性与二酯类相当,氢调敏感性更优。用二醚类催化剂可以制备出具有高熔体流动速率、窄相对分子质量分布的聚丙烯产品,适合于薄壁注塑、纺丝和热压黏合等,也可用于制备灰分极少的电容级聚丙烯薄膜。以琥珀酸酯为内给电子体的催化剂具有良好的立构规整度可控性能(从中到高)[7],可制备宽相对分子质量分布、高刚性的聚丙烯产品,也可用于制备双向拉伸聚丙烯(BOPP)薄膜。
由于上述邻苯二甲酸二酯类化合物属于典型的“塑化剂”,它们对人体的危害引起越来越多的关注,因此,寻找邻苯二甲酸二酯类替代物的呼声也越来越高。近年来,非“塑化剂”内给电子体不断涌现,其中,二醇酯类[8]、磺酰基类[9]和卤代丙二酸酯类[10]等化合物得到了很好的工业应用。
有关Ziegler-Natta催化剂中内给电子体作用的研究很多[11],典型的例子是Correa等[12]利用密度泛函理论计算了硅烷、1,3-二醚、邻苯二甲酸二酯和琥珀酸二酯等四种内给电子体与MgCl2的作用。计算结果显示,前两种化合物由于两个氧原子距离较近,更倾向于与MgCl2的(110)晶面上同一个Mg原子配位;而后两种化合物由于氧原子距离较远,有多种可能的配位方式,与MgCl2(110)和(100)晶面上的Mg原子均可作用,既可与一个Mg原子配位,也可与两个Mg原子配位。进一步的计算还发现,Ti原子周边的琥珀酸酯内给电子体能显著提高Ti活性中心的立体定向能力。
1.1.5 Ziegler-Natta催化体系中的外给电子体
在烯烃的配位聚合中,为了提高聚丙烯的等规度,在聚合反应时往往要加入另一种给电子体,这就是通称的外给电子体。实验结果表明,内、外给电子体要匹配使用。以邻苯二甲酸二酯为内给电子体的催化剂必须与硅烷类外给电子体(如二烷基二甲氧基硅烷)配合使用,才能产生最好的效果。新型的氨基硅烷类外给电子体也有发展,可以在不降低聚合物等规度的情况下提高相对分子质量分布,生产出性能更优异的聚烯烃产品[13]。本课题组也合成了几种新型的含N原子的氨基硅烷类化合物[14],它们是双哌啶二甲氧基硅烷(Donor-Py)、双吗啉二甲氧基硅烷(Donor-Pm)、双1-甲基哌嗪二甲氧基硅烷(Donor-Pz)和双异丙基哌嗪二甲氧基硅烷(Donor-Pi)。将这些化合物作为丙烯聚合用的外给电子体,与工业上常用的外给电子体环己基甲基二甲氧基硅烷(Donor-C)相比,含N原子的氨基硅氧烷类化合物对催化剂的活性、聚合物等规度、相对分子质量分布、氢调敏感性和聚合物的等规序列长度及分布都产生重要的影响。
为了更好地调节烯烃聚合物的结构和性能,使用复配的外给电子体更有效。国内外很多石化企业都开展了这方面的研究,本课题组也利用外给电子体的复配技术制备了高性能聚丙烯树脂[15-16],并与中国石化北京燕山分公司合作,开发出氢调法制备高熔融指数聚丙烯技术,制备了包括高流动均聚物、无规共聚物和抗冲共聚物在内的系列聚丙烯专用料产品,熔融指数(10 min)为20~120 g,主要用于汽车、家居和食品卫生等领域。
研究发现,具有不同烷基和烷氧基取代的硅烷化合物用于丙烯聚合时,生成的聚合物具有不同的性能[17]。这与硅原子相连的烷氧基数目、大小以及与硅原子相连的烃基大小密切相关。烷氧基的数目越多,选择性毒化无规活性中心的能力越强。这是因为一个烷氧基上的氧会与助催化剂AlEt3进行配位,形成复合物;如果硅烷含有一个以上的烷氧基时,复合物中还存在的未配位烷氧基可以选择性地毒化无规活性中心。此外,如果烷氧基太大,可能阻碍复合物与无规活性中心配位,因此,甲氧基硅烷和乙氧基硅烷最常用,而丙氧基硅烷在聚合中的表现较差。虽然不同的研究人员对外给电子体与催化剂活性中心的作用有着不同的观点,但总体来说,大家均认为,在Ziegler-Natta催化体系中存在多种活性中心,如无规活性中心和等规活性中心,其中,等规活性中心根据不同的等规度又分为低等规活性中心、中等规活性中心、次高等规活性中心及高等规活性中心。用于丙烯聚合时,烷氧基硅烷类外给电子体对催化剂活性中心的影响概括起来就是毒化无规活性中心、活化等规活性中心、促进无规活性中心向等规活性中心转变。本课题组的研究工作[18]也支持这种观点。
有关加入外给电子体影响聚合物性能的研究工作也有很多报道。张天一等[19]研究了四种外给电子体Donor-C,Donor-D,Donor-I和Donor-B对聚丙烯性能的影响。实验结果表明,以Donor-D和Donor-I为外给电子体时,制备的聚丙烯等规度较高,相对分子质量分布较宽;以Donor-C为外给电子体时,催化剂具有更敏感的氢调敏感性。本课题组利用SSA技术详细研究了外给电子体Donor-C含量的变化对聚丙烯分子链中各等规组分长度及其分布的影响[20]。实验结果表明,当不添加Donor-C时,制备的聚丙烯经SSA热分级后高等规组分的含量相对较低,而低等规组分的含量相对较高,等规序列长度的分布较窄;随着Donor-C含量的增加,制备的聚丙烯的等规度逐渐增大,经SSA热分级后,聚丙烯分子链中的高等规组分含量也随Si/Ti摩尔比的增加而增加,聚丙烯的等规序列分布也逐渐变宽。
1.2 茂金属催化剂
德国科学家Kamisky等首先发现微量水对二氯二茂锆/AlMe3催化体系有巨大的促进作用,从而采用甲基铝氧烷(MAO)作为茂金属化合物特有的活化剂,使得茂金属催化剂迅速发展起来[21]。
1.2.1 助催化剂MAO
茂金属催化剂与传统的Ziegler-Natta催化剂不同,所用的助催化剂不是通常的烷基铝,而是烷基铝与H2O的反应产物MAO[22]。当然,在通常情况下烷基铝遇H2O就要爆炸,但只要控制好反应条件,就能得到需要的产物。合成MAO最常用的方法是将带有结晶水的无机盐(如CuSO4·5H2O,FeSO4·7H2O,MgSO4·7H2O,Al2(SO4)3·18H2O,MgCl2·6H2O等)作为水源,在控制条件下与AlMe3反应。反应常常在有机溶剂(如甲苯)中进行。低温下将AlMe3缓缓加入到水合盐的悬浮液中,然后控制反应温度和周期,达到一定程度的水解。使用最多的是Al2(SO4)3·18H2O,由于它的结晶水相对含量高,反应残渣少,产率可达60%以上。MAO是齐聚线型链和二维环形结构的混合物,相对分子质量在1 000~1 500范围。
1.2.2 茂金属催化剂的分子设计和聚合物的分子剪裁
对化学家来说,能按照人们的意愿合成结构明确、性能一定的化合物是梦寐以求的事,而茂金属催化剂的出现则为此创造了一个良好的机会。通过控制茂金属催化剂的结构来控制所得聚合物的各种参数,从而达到预期的性能,这就实现了真正意义上的分子剪裁[23]。用于烯烃聚合的茂金属化合物按其结构对称性可划分为五种基本类型:具有C2v对称的茂金属化合物,如Cp2ZrCl2为非手性的,由它生成的聚合物是无规的;外消旋的[En(Ind)2]ZrCl2及其四氢衍生物是C2对称的,其手性特征可用作丙烯的等规聚合;内消旋的En(Ind)2ZrCl2只有一个镜面,无其他对称元素,也只能生成无规聚合物;Me2C(Cp)(Flu)ZrCl2为Cs对称,能生成间规聚合物;无任何对称性的Me2C(MeCp)(Flu)ZrCl2生成的聚合物无明确规律,有时能生成半等规物。
1.2.3 茂金属催化剂用于催化烯烃的共聚合反应
多年的研究和实践使人们慢慢达成一个共识,即无需用茂金属催化剂来全部替代目前工业上通用的Ziegler-Natta催化剂,但是,对制备一些具有特殊性能的聚烯烃专用树脂,则必须使用性能优异的茂金属催化剂。这是因为茂金属催化剂特别有利于烯烃的共聚合反应,而且茂金属催化剂的配体对共聚合行为的影响颇大[24]:使用C2对称的茂金属催化剂能得到无规共聚物;使用Cs对称的催化剂,产物倾向于交替共聚;而采用C1对称的催化剂,产物为交替或嵌段共聚物。因此,在某些特殊的应用领域,茂金属催化剂具有相当大的竞争力。例如,利用茂金属催化剂制备的乙烯/高碳α-烯烃(如乙烯/1-辛烯共聚物)弹性体已被广泛使用,在聚烯烃产品有很好的市场份额。
1.2.4 茂金属聚烯烃催化剂的负载化
使用均相茂金属催化剂得到的聚合物颗粒形态差,往往产生黏釜和团聚现象,对实现工业化生产是不利的。另外,均相茂金属催化剂体系使用MAO的量大,成本高,也不利于工业化生产,解决的办法是均相茂金属催化剂的负载化[25]。
比较了多种无机和有机载体后,硅胶最适宜用作均相茂金属化合物的载体[26]。目前最常用的硅胶负载主要有四种方法。第一种是茂金属化合物直接负载在硅胶上,随后用MAO处理,机理是通过茂金属化合物的锆原子与硅胶表面羟基发生反应,生成锆—氧共价键,在随后加入的MAO的作用下,锆—氧键断裂生成活性中心。第二种是先用MAO处理硅胶等载体,然后与茂金属化合物反应。这时,MAO的Al原子与载体表面的羟基发生反应,茂金属化合物与载体表面的MAO配位,生成载体催化剂。第三种方法是茂金属化合物的配体先与硅胶等载体反应,然后与金属化合物反应,生成茂金属载体催化剂。第四种方法是硅胶等载体先与BF3、SiCl4等表面处理剂反应,然后与MAO作用,最后与茂金属化合物反应得到茂金属载体催化剂。本课题组用硅胶作为载体,相继研究了载体催化剂的制备条件以及各种制备条件对负载催化剂性能的影响,并通过聚合实验筛选出最佳的负载化方案[27]。负载茂金属催化剂用于乙烯聚合的实验结果表明,MAO的加入量大大减少,Al/ Zr摩尔比从原来的2 500降到500以下,聚合物的表观密度提高了3倍。负载茂金属催化剂用于乙烯与己烯的共聚结果表明,它们具有很好的共聚合性能,且控制共聚单体的加入量可以得到中密度、低密度和超低密度的聚合物,以适应不同加工应用的要求。
1.2.5 茂金属催化剂的工业应用
美国的Exxon公司在20世纪90年代初首先实现了茂金属催化乙烯聚合的工业化生产[28]。茂金属聚乙烯的主要产品为线型低密度聚乙烯(LLDPE),是乙烯与α-烯烃的共聚物,用于薄膜、医疗器件及电线电缆等,牌号有上百种。近年来,茂金属聚乙烯在管材上的应用也得到很大的发展。茂金属聚丙烯可分为等规(ipp)、间规(spp)和无规(app)三种聚合物,尤其要指出的是,只有茂金属催化剂才能制备spp。Atofina公司在2002年首先实现了茂金属spp的工业化生产[29],将其命名为Finaplas系列,该系列产品能与热塑性聚烯烃及其他聚合物共混,开发出的新产品具有高熔点、高薄膜收缩率、高透明的特点,并能改进容器成型的加工性能。近年来,我国茂金属催化剂的工业化进程也在提速,中国石化和中国石油两大石化公司都开发出了自己的茂金属聚烯烃工业化产品。
1.3 非茂过渡金属催化剂
非茂过渡金属催化剂的配体不含环戊二烯基团,中心金属与含有未共享电子对的原子(如N,O,P,S等)配位。根据中心金属在元素周期表中的位置不同,又可以分为非茂后过渡金属催化剂[30]和非茂前过渡金属催化剂[31]。
非茂后过渡金属催化剂的一个显著特征是中心金属原子亲电性弱,耐杂原子的能力强,从而使烯烃可能与极性单体共聚合。除此以外,非茂后过渡金属催化剂还能催化环烯烃开环聚合、非环双烯烃易位聚合以及乙烯与一氧化碳的共聚合等。亚胺配位的Pd、Ni催化剂制备的聚乙烯往往具有很高的支化度,产物呈现弹性体性能。另外,吡啶二亚胺配位的Fe齐聚催化剂能制备出C4~C28分布的乙烯齐聚物;而吡啶二亚胺配位的Fe聚合催化剂能制备相对分子质量分布很宽的聚乙烯产品。关于非茂后过渡金属催化剂催化乙烯齐聚与聚合的最新进展,可参阅文献[32]。
非茂前过渡金属催化剂有二齿N为配体的Ti催化剂、二齿酚(O)为配体和三齿(NON)为配体的Ti催化剂,它们有很大的配位空间,对高碳α-烯烃具有很高的活性。β-二酮配位的Ti催化剂对乙烯聚合及乙烯齐聚都表现出良好的活性,该催化剂负载后对丙烯聚合还表现出高的全同立构定向性能。以水杨醛亚胺配位的Ti、Zr催化剂对乙烯配位聚合反应显示出超高的活性[33]。
非茂过渡金属催化剂某些特性是上述Ziegler-Natta催化剂和茂金属催化剂不具备的,加上该催化剂易制备,成本较低,为产业化应用提供了良好契机。
2 烯烃配位聚合反应
如前所述,烯烃配位聚合催化剂的研发是有机金属化学家和聚合物化学家共同努力的结果。后来,物理化学家的加入使对催化剂结构、聚合物链结构与聚合物宏观性能之间关系的研究更深入。研究者通过调控催化剂的结构和聚合条件,就能直接制备出所需的结构新颖、性能优异的聚合物树脂。例如,随着茂金属和非茂金属催化剂的发展,设计合成了各种新型烯烃聚合催化剂,并通过烯烃的共聚合反应,在非极性的聚烯烃树脂中引入极性的功能基团,从而提高了聚烯烃树脂的极性、印染性及与其他材料(如金属、陶瓷、功能聚合物等)的相容性等[34]。
2.1 功能化聚烯烃的制备
如何在聚烯烃链上引入极性基团或极性聚合物链,这是聚烯烃研究领域的重点课题之一。由于新型配位聚合催化剂的不断涌现,能适应极性单体存在的配位聚合反应也取得了一些可喜的结果。
2.1.1 直接共聚合反应
利用配位聚合催化剂,催化烯烃与极性单体的直接共聚合反应,得到的功能化聚烯烃结构可控,方法简单又经济,是最理想的途径。但长期以来困扰人们的问题是催化剂在极性单体存在下迅速失活。Kesti等[35]用B(C6F5)3或[HNMe2Ph] + [B(C6H5)4]-作为助催化剂,活化茂金属催化剂Cp*2ZrMe2和rac-Et(THI)2ZrMe2,研究了烯烃与多种极性单体的共聚合反应,开创了解决催化剂迅速失活问题的新途径。其后,大量的实验结果表明,通过使用过量MAO的方法,或用大位阻烷基铝保护极性基团,或将极性基团远离双键,或使用较大位阻的催化剂等,均能有效抑制催化剂的失活。
中性水杨醛亚胺后过渡金属催化剂用于烯烃和极性单体共聚合反应是由Younkin等[36]首先提出的。该类催化剂具有很强的耐极性单体能力,在乙醇、丙酮、乙醚、三乙胺和水等存在下,能够保持一定的活性。本课题组曾使用非茂前过渡金属催化剂催化乙烯与极性单体共聚合,发现催化剂呈现出较好的耐杂原子能力,活性下降较少[37]。近几年,直接共聚合反应制备功能化聚烯烃的方法发展很快,已有工业化产品上市。
2.1.2 通过反应性单体的制备方法
有一类基团称为反应性基团,如C==C双键、C—B键、C—Si键及苄基等,它们的极性较弱,不会对催化剂进行亲核攻击使催化剂失活,而且它们具有较高的反应活性,在比较温和的条件下可以转化为极性基团。如果将烯烃和含有反应性基团的单体进行共聚合反应,首先制备出含有反应性基团的共聚物,再经过化学改性,将反应性基团转化为极性基团,制备出含极性单体的共聚物;或者转化为聚合引发基团,随之引发其他类单体的聚合反应,制备出聚烯烃接枝共聚物。利用反应性单体法可以制备出含有各种功能基团(如OH—,COOH—,NH2—等)的聚烯烃。在使用反应性单体法制备聚烯烃接枝共聚物时,控制反应性单体在共聚物中的含量就能控制接枝共聚物的接枝点密度;另外,因反应性基团可引发活性聚合,支链长度也可控。因此,反应性单体法的最大优点是能制备出结构可控的聚烯烃接枝共聚物。
本课题组设计了一种新的反应性单体——对烯丙基甲苯,用于替代原有的反应性单体硼烷和对甲基苯乙烯[38],不仅克服了硼烷单体成本太高难于实现工业化的缺点,也解决了对甲基苯乙烯与烯烃不易共聚的难题,具有潜在的工业应用价值。
2.2 原位共聚合反应制备LLDPE
原位共聚合反应是同时使用两种催化剂(双功能催化体系),一种为乙烯齐聚催化剂,另一种为共聚催化剂,反应生成的乙烯齐聚物原位参与共聚合反应,最终产物为LLDPE。本课题组长期从事烯烃原位共聚合反应的研究,使用的双功能催化体系包括以下三种:Ziegler-Natta催化剂加Zieler-Natta催化剂[39];Ziegler-Natta催化剂加茂金属催化剂[40];非茂金属催化剂加茂金属催化剂[41]。这个时间顺序正符合催化剂的发展历程。
我们的研究结果表明,在Ziegler-Natta催化剂加Ziegler-Natta催化剂的组合中,由于齐聚催化剂活性较低,增加齐聚催化剂的用量后使总体活性下降,可能造成催化剂的失活。在Ziegler-Natta催化剂加茂金属催化剂的组合中,前者使用三乙基铝为助催化剂,后者使用MAO为助催化剂,由于两种助催化剂对主催化剂的作用不同,在整个反应体系中就存在互相干扰,影响了催化剂活性的正常发挥。最理想的是非茂金属催化剂加茂金属催化剂的组合,在这种双功能催化体系中,使用的助催化剂都是MAO,具有良好的匹配性,因此重点介绍一下这个体系。
2.2.1 非茂后过渡金属加茂金属组成的双功能催化体系
Brookhart和Gibson各自在研究中发现,吡啶二亚胺类铁(钴)催化剂对乙烯齐聚具有高活性,且对α-烯烃具有高选择性,这为本课题组提出新的乙烯原位共聚双功能催化体系提供了一个良好的机遇[42]。我们以{[2,6-(2-CH3-C6H4N==CCH3)2C5H3N]FeCl2}为齐聚催化剂、rac-Et(Ind)2ZrCl2为共聚催化剂、MAO为助催化剂,组成了双功能催化体系;用乙烯为唯一单体,成功实施了乙烯原位共聚,制备了LLDPE[40]。在该催化体系中,由于使用MAO为唯一助催化剂,使得双功能催化体系中齐聚和共聚两种催化剂之间没有相互干扰,催化活性很高,可达到107g/(mol·h)。聚合物的熔点最低达到84.9 ℃,聚合物的结晶度远低于普通线型聚乙烯的结晶度。除了铁系催化剂外,本课题组还以二亚胺钴化合物作为双功能催化体系的齐聚催化剂,与茂金属催化剂匹配,用于制备LLDPE[41]。
由于齐聚反应得到的是一个宽分布的α-烯烃,而高碳α-烯烃不能参与进一步的共聚合反应,因此,在原位聚合生成的LLDPE中有低聚物残留,影响了共聚物的性能。为了提高共聚物的性能,我们设计合成了新型结构的吡啶二亚胺类铁化合物[2,6-(2,6-F2-C6H3N==CCH3)2C5H3N]FeCl2作为乙烯齐聚催化剂,使结果大有改善[43]。因为新型齐聚催化剂得到的齐聚物α-烯烃的碳数分布窄,且集中分布在低碳数部分,共聚物中就不再出现低聚物残留。
2.2.2 双功能催化体系的负载和工业化
为适应工业生产的需要,均相双功能催化剂体系的负载是十分必要的。除了选用常用的SiO2作为载体外,我们还深入研究了蒙脱土(MMT)载体[44],发现采用MMT负载双功能催化体系后,催化活性仍保持在106g/(mol·h)的水平,得到的乙烯原位共聚物LLDPE的表观密度可达0.35 g/cm3以上,符合工业化生产的要求。
为了实现工业化生产,除了采用MMT负载以外,通过调节Fe催化剂苯环上R1取代基的体积,可以调控生成的齐聚物的碳数分布,进而控制共聚物中支链长度的分布;通过调节Fe/Zr(齐聚催化剂/共聚催化剂)比例,可以控制共聚物中支化单元的含量。用该方法制备的LLDPE既有长支链又有短支链,兼具长支链LLDPE和短支链LLDPE的优点,适于用作制备大型中空容器和管材等。
2.3 链穿梭聚合反应制备烯烃嵌段共聚物
链穿梭(Chain Shuttling)聚合反应是近几年发展起来的一种新的配位聚合反应,可用于制备烯烃嵌段共聚物。与其他聚合反应相比,该反应在制备嵌段共聚物方面有很多优点,容易实现大规模的应用,因此,自2006年美国Dow化学公司几位科学家提出链穿梭聚合的概念以来[45],立刻引起科学界和工业界的广泛注意,目前市场已有性价比很高的烯烃嵌段共聚物销售。
2.3.1 链穿梭聚合反应的基本原理
链穿梭聚合至少采用两种均相烯烃聚合催化剂,并至少使用一种链穿梭剂。在溶液聚合体系中,增长聚合物链从一种催化剂活性中心转移到链转移剂上,再从链转移剂进一步转移到另一种催化剂活性中心上继续增长。这样,以链转移剂为媒介,聚烯烃增长链在多种均相活性中心上不断穿梭,以完成一个聚合物链的增长。链穿梭剂一般是烯烃配位聚合的链转移剂,如二烷基锌、烷基铝等金属有机化合物。因此,链穿梭反应也可看作是多种催化剂和链转移剂组成的可实现交叉链转移的聚合体系。当催化剂的立体选择性或单体选择性不同时,在有效的链穿梭聚合中,可以制备出多嵌段烯烃共聚物。
链穿梭聚合物可以看作是在配位链转移聚合(Coordinative chain-transfer polymerization,CCTP)基础上发展起来的[46]。CCTP是一种特殊的烯烃配位聚合反应。有些烯烃配位聚合催化剂在烷基铝、烷基镁和烷基锌存在下催化烯烃聚合时,在催化剂上生长的聚合物链和烷基金属化合物上的烷基能发生非常快捷的可逆交换,生长的聚合物链转移到烷基金属化合物上,形成休眠的聚合物链,尔后再转移回催化剂活性中心上,使聚合物链继续增长。用这种聚合反应方式制备的聚烯烃相对分子质量分布非常接近于1,有活性聚合的特征。
2.3.2 用链穿梭聚合反应制备烯烃嵌段共聚物
美国Dow化学公司的科学家使用高通量筛选技术,在成百上千的烯烃配位聚合催化剂中寻找到符合链穿梭聚合的催化剂[45],它们分别是双酚氧胺Zr催化剂和吡啶胺Hf催化剂,二乙基锌作为链穿梭剂。他们选择乙烯和1-辛烯作为聚合单体,在上述双催化体系和链穿梭剂的作用下,制备了含有所谓“软”“硬”段的嵌段共聚物。在该反应体系中,Zr催化剂会产生1-辛烯含量较低的“硬”段,而Hf催化剂则易于产生1-辛烯含量较高的“软”段。调节两种催化剂的比例,可以控制“软”“硬”段的比例。
对上述“软”“硬”嵌段共聚物的物理性能进行分析[47],并且与乙烯/1-辛烯无规共聚物相比较,发现该嵌段共聚物具有更高的结晶温度和熔点、更有序的结晶形态和更低的玻璃化转变温度。所以,该嵌段共聚物具有特殊的优良性能,格外受到用户的青睐。
2.4 CCTP反应制备端基功能化聚烯烃
端基功能化聚烯烃(Chain end functionalized polyolefins,Cef-PO)在聚烯烃改性和构筑复杂结构聚合物方面有着重要应用。通过CCTP反应,即聚烯烃链在催化剂金属中心和烷基金属链转移剂之间快速可逆链转移的反应过程,在聚烯烃分子链端基引入双键或简单的功能基团,而后通过基团转化反应得到广泛多样的Cef-PO。在烯烃CCTP反应中,链转移反应有自发的β-H消除或β-Me消除,有向聚合单体或外加链转移剂转移,这些反应都可以用来制备Cef-PO。在烯烃CCTP过程中,链转移反应的类型和可控性等均与使用的催化剂的结构、聚合温度和压力等条件密切相关。因此,要得到期望结构的Cef-PO,必须对上述条件进行精确控制。本课题组多年来在CCTP制备Cef-PO方面作过深入研究,得到了一些可喜的结果[48]。
根据外加链转移剂的数量,通过CCTP反应制备Cef-PO大致可以分为三类[49]:不外加链转移剂的端双键聚烯烃的制备及功能化;外加一种链转移剂的原位制备Cef-PO;外加两种链转移剂的连续链转移过程。
最近,本课题组以亚胺铁为催化剂,高活性、高选择性地制备出乙烯基封端聚乙烯(v-PE),并经油剂抽提分级处理得到低相对分子质量、窄相对分子质量分布的v-PE[50]。进一步利用环氧基团开环和硫基-烯烃点击化学反应,制备出羟基、卤素、叠氮、氨基、磺酸基、羧基、三甲基硅烷及受阻酚基团封端的聚乙烯。此外,环氧基团封端聚乙烯在碱性条件下经聚乙二醇单甲醚处理,可以直接得到聚乙烯-聚乙二醇嵌段共聚物。研究发现,该嵌段共聚物具有良好的亲水性和亲油性,而所采用的反应具有温和、易操作及选择性高等特点。
3 结语
烯烃的配位聚合从催化剂到聚合反应、工艺和产品研究,最后落实到高性能聚烯烃材料的开发和应用,一路走来,经过六十余年,终于得到了今天如此辉煌的业绩,这是几代科学研究和工程技术人员共同努力的结果。聚烯烃工业的发展史给人们提供了很多启发,不断的创新才能推动科学技术的进步。目前,有关烯烃配位聚合反应的机理问题还没有得到很好的解决,新的催化剂和新的聚合物还在不断涌现。总之,新问题还会不断出现,但这正符合了科学发展的规律。
最后对烯烃配位聚合提出几点认识和建议:
1)要充分认识过渡金属催化剂是促进聚烯烃工业进步的关键技术。无论是最早发现但至今仍很出彩的Ziegler-Natta催化剂,还是后续发展并将逐步发挥工业应用价值的茂金属和非茂金属催化剂,无一例外。可以说,没有催化剂的发展就没有聚烯烃工业的今天。
2)随着经济的发展和成熟,聚烯烃工业进入了市场主导或成本主导时期。因此,使用什么催化剂,生产什么产品,应由市场来决定。由于Ziegler-Natta催化剂使用起来非常经济,近几年来它的性能还通过使用外给电子体及其复合技术而得到很大提高,因此,Ziegler-Natta催化剂目前在聚烯烃工业上仍占主导地位。尽管如此,由于茂金属和非茂金属催化剂具有制备新型特种树脂的优点,因此,随着工业的发展和社会需求的升级,茂金属和非茂金属催化剂占主导地位已为时不远,对此一定要有充分的认识。
3)实现催化剂和聚烯烃材料的高性能定制生产。由于催化剂制造是一个技术集约型和高成本的产业,因此,由一些专注于催化剂制备的公司为聚烯烃厂家定制生产催化剂,或授权生产新开发的催化剂,是工业发展的必然趋势。这样,催化剂授权者可集中精力和资源专注于聚合物制备,成本低、效益高。对于茂金属和非茂金属催化剂的制备,由于催化剂制造公司不仅要生产出活性良好的配位化合物,还要实现高效的化合物负载化,这种定制生产方式更显得重要。随着茂金属和非茂金属催化剂的迅速发展,有很多长期以来存在的美好愿望都可以实现。例如,聚烯烃的相对分子质量及其分布的精确调节;聚合物链结构中长支链和短支链、嵌段和无规分布的人为控制;共聚单体中极性和非极性的任意选择;立体化学结构中的等规、间规和无规产品的定量安排等。聚烯烃生产者要根据客户提出的产品性能要求进行定制生产,这是从事烯烃配位聚合工作者们新的责任。
[1] 周倩,秦亚伟,李化毅,等. Ziegler-Natta催化剂的载体技术研究进展[J].高分子通报,2016(9):126-139.
[2] Hu Youliang,Chien J C W. Superactive and stereospecific catalysts.Ⅰ. Structure and productivity[J].J Polym Sci Part A:Polym Chem,1988,26(8):2003-2018.
[3] Luo Zhi,Zheng Tao,Li Huayi,et al. A submicron spherical polypropylene prepared by heterogeneous Ziegler-Natta catalyst[J].Ind Eng Chem Res,2015,54(44):11247-11250.
[4] Toho Titanium Co.,Ltd. Catalyst for the polymerization of olefins:US4640906[P].1987-02-03.
[5] Shell Oil Company. Olefin polymerization catalyst composition:US4728705[P].1988-03-01.
[6] Himont Incorporated. Components and catalysts for the polymerization of olefins:US4971937[P].1990-11-20.
[7] Montell Technology Company. Catalyst composition for the copolymerization of propylene:WO00/63261[P].2000-10-26.
[8] 中国石油化工股份有限公司. 用于烯烃聚合反应的催化剂组分及其催化剂:03109781. 2[P].2003-04-21.
[9] 中国石油天然气股份有限公司. 一种烯烃聚合催化剂及其制备方法和应用:200710118854 X[P].2007-06-13.
[10] 陶氏环球技术有限公司. 具有卤代-丙二酸酯内给电子体的催化剂组合物和由其制备的聚合物:201180068102.1[P].2011-12-02.
[11] 胡友良,常贺飞,李化毅,等. 内外给电子体在丙烯聚合Ziegler-Natta催化体系中作用的研究进展(上) [J].石油化工,2013,46(11):1189-1196.
[12] Correa A,Piemontesi F,Morini G,et al. Key elements in the structure and function relationship of the MgCl2/TiCl4/Lewis base Ziegler-Natta catalytic system[J].Macromolecules,2007,40(25):9181-9189.
[13] Equistar Chemicals. LP. Methods for preparing propylene polymer having broad molecular weight distribution:US6800703 B1[P].2004-11-05.
[14] 中国科学院化学研究所. 一种含N硅烷类化合物的制备方法及其用于丙烯聚合的应用:201210387272. 2[P].2012-10-15.
[15] 中国科学院化学研究所. 氢调法制备高熔融指数聚丙烯的催化剂及聚合方法:201110097918[P].2011-04-19.
[16] Chang Hefei,Ren Shitong,Dang Xiaofei,et al. The effect of the mixed external donors on the sequence length distribution of polypropylene[J].J Appl Polym Sci,2013,129(3):1026-1035.
[17] Seppälä J V,Härkönen M,Luciani L. Effect of the external alkoxysilane donors on the polymerization of propylene with high activity Ziegler-Natta catalysts[J].Die Makromol Chem,1989,190(10):2535-2550.
[18] 李化毅,张辽云,胡友良. 密度泛函理论研究聚烯烃催化剂取代基电子效应与催化活性的关系[J].催化学报,2010,31(9):1127-1131.
[19] 张天一,夏先知,刘月祥,等. 外给电子体对DQC催化剂聚合性能的影响[J].石油化工,2011,40(4):381-386.
[20] Chang Hefei,Zhang Yu,Ren Shitong,et al. Study on the sequence length distribution of polypropylene by the successiveself-nucleation and annealing(SSA) calorimetric technique[J].Polym Chem,2012,3(10):2909-2919.
[21] Kaminsky W,Miri M. Ethylene propylene diene terpolymers produced with a homogeneous and highly active zirconium catalyst[J].Polym Sci Polym Ed,1985,23(8):2151-2164.
[22] 黄葆同,陈伟. 茂金属催化剂及其烯烃聚合物[M].北京:化学工业出版社,2000:7-10.
[23] Ressoni L,Piemontest F. Olefin polymerization at bis(pentamethy cyclo-pentadienyl)-zirconium and -hafnium centers:Chain-transfer mechanisms[J].J Am Chem Soc,1992,114(3):1025-1032.
[24] Herfert N,Montag P,Fink G. Elementary processes of Ziegler-Catalysis. 7. Ethylene,α-olefin and norbornene copolymerization with the stereorigid catalyst systems iPr[FluCp]-ZrCl2/MAO and Me2Si[Ind]2ZrCl2/MAO[J].Makromol Chem Macromol Chem Phys,1993,194(11):3167-3182.
[25] Severn J R,Chadwick J C,Duchateau R,et al. “Bound but not gagged”—Immobilizing single-site α-olefin polymerization catalysts[J].Chem Rev,2005,105(11):4073-4174.
[26] Fink G,Steinmetz B,Zechlin J,et al. Propene polymerization with silica-supported metallocene/MAO catalysts[J]. Chem Rev,2000,100(4):1377-1390.
[27] 胡友良. 烯烃聚合催化剂研究开发进展[J].石化技术与应用,2002,20(1):1-6.
[28] 唐岩,李延亮,王群涛,等. 茂金属催化剂及茂金属聚乙烯现状[J].合成树脂及塑料,2014,31(2):76-80.
[29] Atofina commercializes syndiotactic PP[J].Chem Week,2002,164(11):32.
[30] Rieger B编. 黄葆同,李悦生译. 后过渡金属聚合催化[M].北京:化学工业出版社,2005:9-23.
[31] 陈商涛,胡友良. β-二酮类有机金属配合物催化烯烃配位聚合的研究进展[J].石油化工,2006,35(9):896-902.
[32] 王一清,黄佳兵,张文娟,等. 后过渡金属催化乙烯齐聚与聚合进展[J].高分子通报,2016(9):52-62.
[33] Furuyama R,Fujita T,Funaki S F,et al. New high-performance catalysts developed at Mitsui Chemicals for polyolefins and organic synthesis[J].Cat Sur Asia,2004,8(1):61-71.
[34] 胡友良,乔金樑,吕立新. 聚烯烃功能化及改性——科学与技术[M].北京:化学工业出版社,2006:7-28.
[35] Kesti M R,Coates G W,Waymouth R M. Homogeneous Ziegler-Natta polymerization of functionalized monomers catalyzed by cationic group Ⅳ metallocenes[J].J Am Chem Soc,1992,114(24):9679-9978.
[36] Younkin T R,Conner E F,Henderson J I,et al. Neutral,single-component nickel(Ⅱ) polyolefin catalysts that tolerate heteroatoms[J].Science,2000,287(5452):460-462.
[37] Zhang Xiaofan,Chen Shangtao,Li Huayi,et al. Copolymerizations of ethylene and polar comonomers with bis(phenoxyketimine) group Ⅳ complexes:Effects of the central metal properties[J].J Polym Sci Part A:Polym Chem,2007,45(1):59-68.
[38] Li Huayi,Chen Shangtao,Zhang Xiaofan,et al. Synthesis and functionalization of ethylene/p-allyltoluene copolymer[J]. Reac Funct Polym,2005,65(3):285-292.
[39] 谢光华,方义群. 双催化剂体系乙烯二聚和共聚合的研究[J].石油化工,1994,23(4):191-197.
[40] Liu Zhongyang,Xu Demin,Guo Cunyue,et al. LLDPE prepared by in situ polymerization using a dual-functional catalytic system[J].Chin Sci Bull,2001,46(24):2051-2054.
[41] Wang Hang,Ma Zhi,Ke Yucai,et al. Synthesis of linear low density polyethylene(LLDPE) by in situ copolymerization with novel cobalt and zirconium catalysts[J].Polym Int,2003,52(9):1546-1552.
[42] 柳忠阳,王军,徐德民,等. 后过渡金属及茂金属组成的催化剂体系原位聚合制备长链支化聚乙烯[J].科学通报,2001,46(15):195-198.
[43] Zhang Zhicheng,Cui Nannan,Lu Yingying,et al. Preparation of linear low-density polyethylene by the in situ copolymerization of ethylene with an iron oligomerization catalyst and rac-ethylene bis(indenyl) zirconium(Ⅳ) dichloride[J].J Polym Sci Part A:Polym Chem,2005,43(5):984-993.
[44] 中国科学院化学研究所. 一种蒙脱土载体化双功能催化体系及制备方法和应用:03110318. 9[P].2003-04-08.
[45] Arriola D J,Carnahan E M,Hustad P D,et al. Catalytic production of olefin block copolymers via chain shuttling polymerization[J].Science,2006,312(5774):714-719.
[46] Kemp R. How to polymerize ethylene in a highly controlled fashion?[J].Chem Eur J,2007,13(10):2764-2773.
[47] Khariwala D U,Taha A,Chum S P,et al. Cristallization kinetics of some new olefinic block copolymers[J].Polymer,2008,49(5):1365-137 5.
[48] 张勇杰,李化毅,董金勇,等. 端基功能化聚烯烃的合成与应用[J].化学进展,2014,26(1):110-124.
[49] Dong Jinyong,Hu Youliang. Design and synthesis of structurally well-defined functional polyolefins via transition metalmediated olefin polymerization chemistry[J].Coord Chem Rev,2006,250(1/2):47-65.
[50] Zhang Yongjie,Li Huayi,Dong Jinyong,et al. Facile synthesis of chain end functionalized polyethylenes via epoxide ring-opening and thiol-ene addition click chemistry[J].Polym Chem,2014,5(1):105-115.
(编辑 王 萍)
Catalysts for olefin coordination polymerization and polymerization reaction
Hu Youliang
(Key Laboratory of Engineering Plastics,Institute of Chemistry,Chinese Academy of Sciences,Beijing 100190,China)
In this paper,catalysts for olefin coordination polymerization and polymerization reaction are reviewed. Researches in our group relevant to this subject are also referenced. The Ziegler-Natta catalysts,metallocene and non-metallocene catalysts are discussed. The synthesis of functional polyolefin,preparation of linear low-density polyethylene by in situ copolymerization of ethylene,preparation of polyolefin block copolymer by chain shuttling polymerization,and synthesis of chain end functionalized polyolefin are introduced. Some suggestions are given for research and development of polyolefin.
polyolefin;coordination polymerization;Ziegler-Natta catalysts;metallocene catalysts;non-metallocene catalysts
1000-8144(2017)06-0651-09
TQ 325.1
A
10.3969/j.issn.1000-8144.2017.06.001
2017-02-17;[修改稿日期]2017-04-17。
胡友良(1942—),男,江苏省无锡市人,大学,研究员,电话 010-62562815,电邮 huyl@iccas.ac.cn。
