盆底器官脱垂患者子宫骶韧带组织LC3Ⅱ、COLⅠ和COL Ⅲ的表达变化
2017-06-05王红南方
王红,南方
(鄂州市中心医院,湖北鄂州436099)
盆底器官脱垂患者子宫骶韧带组织LC3Ⅱ、COLⅠ和COL Ⅲ的表达变化
王红,南方
(鄂州市中心医院,湖北鄂州436099)
目的 观察盆底器官脱垂(POP)患者子宫骶韧带组织中自噬相关蛋白微管相关蛋白1轻链3Ⅱ(LC3Ⅱ)、Ⅰ型胶原(COLⅠ)和Ⅲ型胶原(COLⅢ)的表达变化,并探讨自噬与POP的关系。方法 轻度POP(POPⅠ~Ⅱ度)14例(A组)、重度POP(POPⅢ~Ⅳ级)17例(B组)、无POP者17例(C组),在子宫全切术术中均留取子宫骶韧带组织,采用Western blotting法检测各组子宫骶韧带组织LC3、COLⅠ和COL Ⅲ。采用Pearson相关性分析方法分析LC3、COLⅠ和COL Ⅲ的相关性。结果 A、B、C组LC3蛋白相对表达量分别为30.41±2.76、24.28±2.61、38.15±2.19,COLⅠ蛋白相对表达量分别为30.41±2.76、24.28±2.61、38.15±2.19,COLⅢ蛋白相对表达量分别为31.50±2.15、26.00±2.10、37.29±2.13,组间两两比较,P均<0.05。Pearson相关性分析显示,轻度POP患者子宫骶韧带组织中LC3Ⅱ与COLⅠ和COLⅢ均呈正相关(r分别为0.736 2,0.781 9;P均<0.01);重度POP患者子宫骶韧带组织中LC3Ⅱ与COLⅠ和COLⅢ均呈正相关(r分别为-0.865,-0.709 4;P均<0.01)。结论 POP患者子宫骶韧带组织LC3、COLⅠ和COL Ⅲ均呈低表达。LC3、COLⅠ和COL Ⅲ可能与POP的发生有关。
盆底器官脱垂;子宫骶韧带;微管相关蛋白1轻链3Ⅱ;Ⅰ型胶原;Ⅲ型胶原
盆底器官脱垂(POP)主要发生在于经产妇[1],发病率较高[2,3]。流行病学调查发现,POP的发病主要与分娩、年龄、肥胖、慢性腹压增加及绝经等因素相关[4]。但目前关于POP发病的具体分子机制仍不明确。自噬是生物体在长期进化中形成的一种自我保护机制,本质上是一种细胞促存活机制。微管相关蛋白1轻链3Ⅱ(LC3Ⅱ)是自噬体膜上的标志性蛋白,Ⅰ型胶原(COLⅠ)和Ⅲ型胶原(COLⅢ)是自噬标志蛋白。目前关于自噬在POP中的作用机制相关报道较少。我们观察了POP患者子宫骶韧带组织中LC3Ⅱ、COLⅠ和COLⅢ的表达变化,并探讨自噬与POP的关系。现将结果报告如下。
1 资料与方法
1.1 临床资料 选择2015年1月~2016年1月间在鄂州市中心医院行子宫全切术的住院患者48例,其中31例因POP行子宫全切术,因子宫腺肌症、子宫肌瘤或功能性子宫出血等良性疾病行子宫全切术者17例;48例患者均为阴道分娩,均已绝经,既往无激素使用史,无明显盆腔炎症,无糖尿病或高血压等疾病,无各种慢性疾病。根据国际尿控协会(ICS)1996年公布的POP-Q分类法将48例患者分为轻度POP(POPⅠ~Ⅱ度)者14例(A组)、重度POP(POPⅢ~Ⅳ级)者17例(B组)、无POP者17例(C组)。A组年龄(52.50±5.49)岁,BMI(22.57±3.35)kg/m2,分娩次数(2.40±1.18)次;B组年龄(51.88±3.90)岁,BMI(22.94±3.29) kg/m2,分娩次数(2.59±1.33)次,C组年龄(51.35±5.57)岁,BMI(22.71±3.18) kg/m2,分娩次数(2.41±1.18)次。三组年龄、阴道分娩次数、BMI等资料具有可比性。
1.2 各组子宫骶韧带组织LC3、COLⅠ和COL Ⅲ检测 三组均行子宫全切术,术中留取距离宫颈约0.5 cm处宫骶韧带组织,长约0.5 cm。采用Western blotting法检测各组子宫骶韧带组织LC3、COLⅠ和COL Ⅲ。取各组组织,加入细胞裂解液充分裂解,离心后取上清,采用BCA试剂盒检测蛋白浓度。配胶(分离胶10%)、上样、电泳、转膜(PVDF膜)、封闭、一抗孵育、二抗孵育。洗膜后ECL显色液显色。方正扫描仪扫描条带,运用Quantity One软件对条带进行灰度分析,以LC3、COLⅠ和COL Ⅲ与GAPDH条带的相对灰度值表示LC3、COLⅠ和COL Ⅲ蛋白的相对表达量。
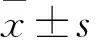
2 结果
三组子宫骶韧带组织LC3、COLⅠ和COL Ⅲ蛋白的相对表达量比较见表1。

表1 三组子宫骶韧带组织LC3、COLⅠ和COL Ⅲ蛋白的相对表达量比较
注:与C组比较,#P<0.05;与B组比较,*P<0.05。
Pearson相关性分析显示,轻度POP患者子宫骶韧带组织中LC3Ⅱ与COLⅠ和COLⅢ均呈正相关(r分别为0.736 2、0.781 9,P均<0.01);重度POP患者子宫骶韧带组织中LC3Ⅱ与COLⅠ和COLⅢ均呈正相关(r分别为-0.865、-0.709 4,P均<0.01)。
3 讨论
女性盆底器官主要是韧带和肌肉固定于相应的位置,以至于不随应力改变位置而影响功能,而子宫主骶韧带是提供力学抗性的关键结构其主要成分是肌肉和胶原纤维。POP患者往往是这些支持结构发生了病变,其内的胶原蛋白等细胞外基质(ECM)成分发生了改变,导致结缔组织松弛,最终引起POP。
自噬作为一种防御机制,在细胞缺氧、饥饿和生长因子缺乏等条件下,以及一些病理状态下,通过对胞内物质的降解和循环利用来改变新陈代谢,使细胞得以生存,对维持细胞的存活有积极作用,是细胞在低氧环境下生存的可能机制,在本质上是一种细胞促存活机制[5],且具有双向作用[6],适度的自噬有利于细胞的生存,过度自噬则会导致细胞死亡。Liu等[7]研究证实,提高自噬水平可抑制ECM的过度聚集,保护心肌细胞,促进胶原降解来抑制纤维化的进程[8]。而另一项风湿性关节炎疾病研究[9, 10]发现,自噬可促进滑膜内纤维母细胞的存活,抑制其凋亡,从而使得关节腔内的ECM过度分泌聚集,对受累关节造成永久性的损伤。自噬过程由一系列自噬相关蛋白(Atg蛋白)介导完成,这些蛋白质在自噬体形成的不同阶段发挥作用。
LC3是自噬体膜上的标记蛋白,细胞内存在两种形式的LC3蛋白:LC3Ⅰ和LC3Ⅱ。LC3蛋白在合成后其C端即被Atg4蛋白酶切割变成LC3Ⅰ,LC3Ⅰ散在分布于细胞浆内,当自噬体形成后,LC3Ⅰ和磷脂酰乙醇胺(PE)偶联形成LC3 Ⅱ并定位于自噬体内膜和外膜。与其他一些定位于自噬性结构膜上的Atg蛋白不同(仅在自噬过程的某一阶段发挥作用),LC3Ⅱ始终稳定地保留在自噬体膜上直到与溶酶体融合,因此被用来作为自噬体的标记,且LC3Ⅱ的水平在某种程度上反映了自噬体的数量[11],是目前最明确贯穿整个自噬过程,并能够出现在晚期自噬溶酶体中的自噬相关蛋白,所以LC3-Ⅱ作为自噬的特异性标志物受到广泛的肯定。本研究发现, A、B组患者子宫骶韧带组织中LC3Ⅱ、COLⅠ和COLⅢ的蛋白的相对表达量低于C组,与Mao等[12]研究成果一致。COLⅠ和COLⅢ是成纤维细胞主要的细胞外基质成分之一,多数学者认为胶原合成的减少,分解的增加,胶原之间交联减少,形态异常,COLⅠ和COLⅢ比例下降,这些都在POP的发生发展中具有重要的作用[13]。
相关性分析发现,轻度POP患者子宫骶韧带组织中LC3Ⅱ与COLⅠ和COLⅢ均呈正相关;重度POP患者子宫骶韧带组织中LC3Ⅱ与COLⅠ和COLⅢ均呈正相关。那么胶原的合成代谢是否受自噬的影响,目前还需更深层次的研究,但本研究结果发现的正相关提示了这种可能。胶原的合成代谢与多种细胞生长因子有关,其中转化生长因子TGF-β1在调节胶原代谢过程中起关键作用,可通过多条途径调节胶原合成和代谢[14]。Ghavami等[15]用TGF-β1诱导原代人心前房肌成纤维细胞发生纤维化,发现TGF-β1能够通过激活细胞发生自噬,促进COLⅠ、纤连蛋白高表达,自噬抑制剂和敲除自噬相关基因均可抑制TGF-β1的促纤维化作用。有学者[16,17]用小鼠胚胎成纤维细胞建立细胞饥饿模型,TGF-β1可缓解细胞饥饿导致的细胞生长受限、线粒体损伤和细胞凋亡,进一步细胞嫁接模型实验发现自噬可增强TGF-β1介导的成纤维细胞保护作用,说明自噬参与了TGF-β1介导的对肿瘤相关成纤维细胞的保护和促增殖过程。
综上所述,POP患者子宫骶韧带组织LC3、COLⅠ和COL Ⅲ均呈低表达。LC3、COLⅠ和COL Ⅲ在POP的发生发展中可能存在相互作用。但其具体作用机制尚有待于进一步研究。
[1] 郎景和,朱兰. 女性盆底学[M]. 人民卫生出版社,2008:315-319.
[2] Weber AM, Richter HE. Pelvic organ prolapse[J]. Obstet Gynecol, 2005,106(3):615-634.
[3] 王静怡,朱兰. 成年女性盆腔脏器脱垂流行病学研究进展[J]. 实用妇产科杂志,2011,27(2):95-98.
[4] Medvedev AE, Piao W, Shoenfelt J, et al. Role of TLR4 tyrosine phosphorylation in signal transduction and endotoxin tolerance[J]. J Biol Chem, 2007,282(22):16042-16053.
[5] Mizushima N. Autophagy: process and function[J]. Genes Dev, 2007,21(22):2861-2873.
[6] Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword[J]. Science, 2004,306(5698):990-995.
[7] Liu S, Chen S, Li M, et al. Autophagy activation attenuates angiotensin II-induced cardiac fibrosis[J]. Arch Biochem Biophys, 2016,2016(590):37-47.
[8] Aranguiz-Urroz P, Canales J, Copaja M, et al. Beta(2)-adrenergic receptor regulates cardiac fibroblast autophagy and collagen degradation[J]. Biochim Biophys Acta, 2011,1812(1):23-31.
[9] Connor AM, Mahomed N, Gandhi R, et al. TNFalpha modulates protein degradation pathways in rheumatoid arthritis synovial fibroblasts[J]. Arthritis Res Ther, 2012,14(2):62.
[10] Shin YJ, Han SH, Kim DS, et al. Autophagy induction and CHOP under-expression promotes survival of fibroblasts from rheumatoid arthritis patients under endoplasmic reticulum stress[J]. Arthritis Res Ther, 2010,12(1):19.
[11] Kimura S, Fujita N, Noda T, et al. Monitoring autophagy in mammalian cultured cells through the dynamics of LC3[J]. Methods Enzymol, 2009,2009(452):1-12.
[12] Tomasek JJ, Gabbiani G, Hinz B, et al. Myofibroblasts and mechano-regulation of connective tissue remodelling[J]. Nat Rev Mol Cell Biol, 2002,3(5):349-363.
[13] Edwall L, Carlstrom K, Fianu JA. Markers of collagen synthesis and degradation in urogenital tissue and serum from women with and without uterovaginal prolapse[J]. Mol Hum Reprod, 2008,14(3):193-197.
[14] Sun Y, Zhu F, Yu X, et al. Treatment of established peritoneal fibrosis by gene transfer of Smad7 in a rat model of peritoneal dialysis[J]. Am J Nephrol, 2009,30(1):84-94.
[15] Ghavami S, Cunnington RH, Gupta S, et al. Autophagy is a regulator of TGF-beta1-induced fibrogenesis in primary human atrial myofibroblasts[J]. Cell Death Dis, 2015,2015(6):1696.
[16] Liu FL, Mo EP, Yang L, et al. Autophagy is involved in TGF-beta1-induced protective mechanisms and formation of cancer-associated fibroblasts phenotype in tumor microenvironment[J]. Oncotarget, 2016,7(4):4122-4141.
[17] 邰海凤,陈姝彤,童晓文.自噬基因 Beclin1在宫颈癌中的研究进展[J].山东医药,2014,2014(9):97-99.
南方(E-mail: 847285973@qq.com)
10.3969/j.issn.1002-266X.2017.19.022
R711.2
B
1002-266X(2017)19-0072-03
2016-11-12)
