骶骨富于细胞神经鞘瘤8例临床病理分析
2017-06-05杨道华华莹奇
林 军,杨道华,华莹奇,陈 安
骶骨富于细胞神经鞘瘤8例临床病理分析
林 军1,杨道华1,华莹奇2,陈 安3
目的 探讨骶骨富于细胞神经鞘瘤的临床病理特点及与骶骨经典型神经鞘瘤的异同。方法 回顾性分析8例发生于骶骨的富于细胞神经鞘瘤的临床资料、影像学、病理学特征、免疫表型、鉴别诊断及预后。结果 8例中女性5例,男性3例,平均年龄46.4岁。临床上以骶尾部疼痛为主,影像学上表现为骶骨或骶骨及骶前肿块。镜下见富于细胞神经鞘瘤由梭形细胞组成,呈条束状、交织状排列,无明显栅栏状排列和“verocay小体”结构,未见明显束状区和网状区,伴有骨质破坏。免疫表型:瘤细胞S-100蛋白和vimentin均弥漫强阳性,Ki-67增殖指数3%~10%。8例中有4例为复发病例,平均复发时间6.5年。结论 骶骨富于细胞神经鞘瘤是一种少见肿瘤,由于其形态学和生长方式与骶骨经典型神经鞘瘤有一定的差异,故病理诊断时应将该肿瘤给予注明,以供临床随访、治疗。
骶骨肿瘤;富于细胞神经鞘瘤;经典型神经鞘瘤;免疫组织化学
骶骨发生的原发性肿瘤属于少见肿瘤,其中又以脊索瘤和骨巨细胞瘤多见,虽然WHO(2013)骨肿瘤分类将神经鞘瘤从分类中删除[1],但由于骶骨及其周围结构的复杂性,临床及影像学往往将该部位发生的神经源性肿瘤视为骶骨区域原发性肿瘤,国内外文献也大多侧重临床和影像学方面的研究和报道。本文回顾性分析8例骶骨富于细胞神经鞘瘤的临床病理学、影像学、免疫表型、鉴别诊断及预后,并复习相关文献,旨在提高对其的认识水平。
1 材料与方法
1.1 材料 收集上海市第一人民医院病理科2014年10月~2016年6月诊断的骶骨富于细胞神经鞘瘤8例,均具有完整资料。另收集同期发生在骶骨区域的经典型神经鞘瘤6例作为对照组。病理切片均经两名有经验的病理医师复查。
1.2 方法 所有标本均经10%中性福尔马林固定、石蜡包埋,HE染色,镜下观察。免疫组化染色采用EnVision法。抗体S-100(1 ∶200)、CD57(1 ∶100)、GFAP(1 ∶400)、vimentin(1 ∶400)、CD34(1 ∶200)、NF(1 ∶100)、Ki-67(1 ∶200)、EMA(1 ∶400)和SMA(1 ∶800)均购自长岛公司。二抗为基因科技(上海)的GTVision抗鼠/兔通用型免疫组化检测试剂盒。操作步骤严格按说明书进行。
2 结果
2.1 临床特征 8例中女性5例,男性3例;发病年龄23~65岁,平均46.4岁。4例为原发肿瘤,4例为复发肿瘤,既往均有相关病史。5例患者发病时有不同程度的骶尾部疼痛不适,1例表现为腰背部疼痛,1例为备孕时检查发现;3例伴有下肢麻木感。影像学上2例表现为骶骨内肿块,骶管扩大,伴骨质破坏;另6例除骶骨内肿块,CT或MRI检查可见骶管扩张、骶管内肿瘤沿神经孔向外生长、同时形成骶前软组织包块(表1)。MRI检查所有肿瘤为均匀一致的信号改变,T1像为等高或稍低信号、T2像为高信号(图1、2)。肿瘤均起源于S3及以上,其中1例累及到S4。2例骶管内肿瘤患者采用的是经前路入口,其余6例有骶前肿块患者采用经前后路入口对肿瘤采用肿瘤边缘性切除。
2.2 眼观 骶骨内肿瘤送检组织均为碎组织,骶前肿块大部分为完整肿块,个别病例为碎组织。肿块直径7~24.5 cm,平均14.1 cm。颜色表现为灰白、灰红、灰黄色。经典型神经鞘瘤的送检组织除2例仅为骶前肿块的病例为完整肿块,其余均为碎组织,肿块直径2.5~13 cm,平均7.2 cm。
2.3 镜检 6例经典型神经鞘瘤有Antoni A区(束状区)和Antoni B区(网状区)结构,以A区为主,可见梭形细胞核呈栅栏状排列,有“verocay小体”结构,均缺乏细胞异型性和核分裂象,部分区域可见纤维性包膜,伴有不同程度的骨质破坏。8例富于细胞神经鞘瘤由梭形细胞组成,呈条束状、交织状排列(图3),无栅栏状排列和“verocay小体”结构,未见明显束状区和网状区。部分区域内,细胞核染色质较粗、深染,并显示一定的多形性(图4),在多数肿瘤中均可见少量的核分裂象(图5),但不见病理性核分裂象,也不见凝固性坏死。肿瘤内血管周围可见少量的淋巴细胞浸润,形成淋巴血管套(图6)。有明显增厚的包膜,内见多少不等的淋巴细胞聚集,形成袖套样结构(图7)。贴近包膜处可见漩涡样结构(图8)。骶骨内肿瘤经常有骨质破坏(图9)。在多数病例中能见到类似经典型神经鞘瘤中的一些形态特点,如在梭形瘤细胞之间可见灶性泡沫细胞聚集(图10),以及伴有明显玻璃样变性的厚壁血管(图11)。
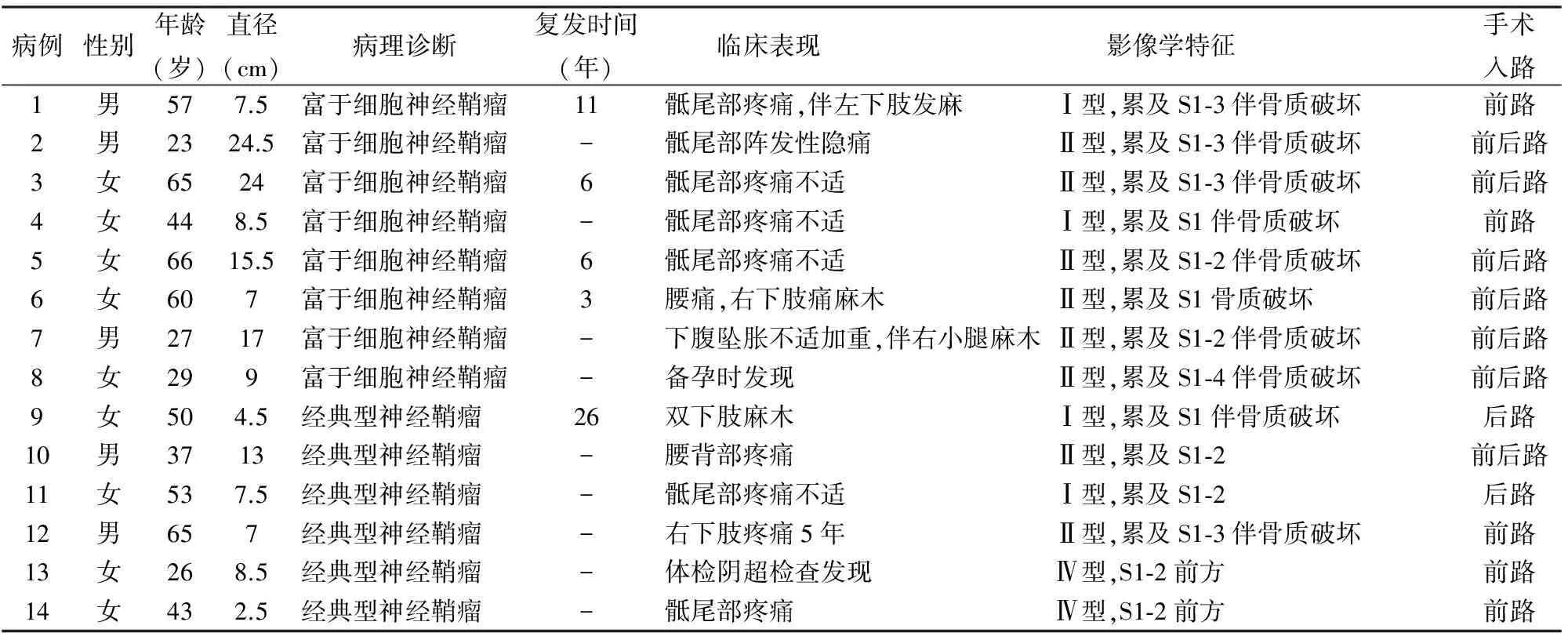
表1 8例骶骨富于细胞神经鞘瘤及6例经典型神经鞘瘤的临床资料
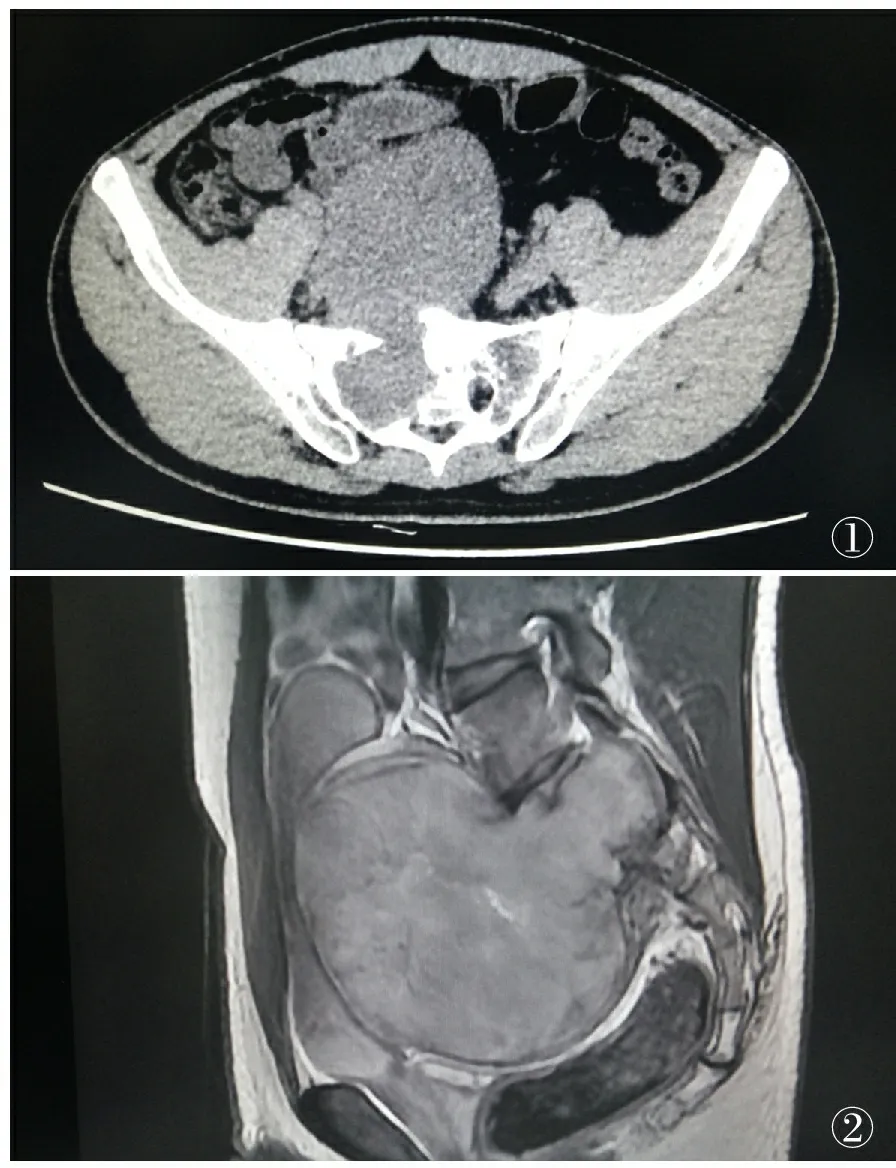
①②
图1 例7:CT示骶骨偏右份骨质破坏并骶前肿块 图2 例7:MRI增强示S1-2椎体破坏,并骶前肿块,T2像高信号,其内信号不均,似“哑铃状”
2.4 免疫表型 瘤细胞S-100蛋白(图12)和vimentin均弥漫强阳性,与经典型神经鞘瘤一致。2例弥漫性表达GFAP、1例弥漫性及1例灶性表达CD57,其余均为阴性,而经典型神经鞘瘤多数灶性表达CD57和GFAP。2例灶性、弱阳性表达EMA,经典型神经鞘瘤均为阴性。其他标志物包括SMA、CD34、NF两组肿瘤均为阴性。8例骶骨富于细胞神经鞘瘤Ki-67增殖指数为3%~10%,平均约6%(复发病例为6.25%,初发病例为5.75%),6例经典型神经鞘瘤为1%~8%,平均约3.5%。
3 讨论
骶骨位置较深,解剖结构复杂,其神经源性肿瘤主要发生在椎管内外的神经根,通过骶孔与骶管相通,可导致骶孔扩大和骨质破坏,典型者表现为“哑铃型”改变。这些肿瘤生长缓慢,初期无明显临床症状,部分患者体检时或怀孕后检查发现。随着肿块的进一步增大,多数患者出现骶尾部的不适、疼痛,多数伴有下肢麻木。按照肿瘤起源及生长特点将骶骨神经源性肿瘤分为四种类型:Ⅰ型,肿瘤生长仅限于骶管内、骶管膨胀扩大;Ⅱ型,肿瘤出现骶神经孔向前生长,形成巨大骶前肿块;Ⅲ型,肿瘤向前、向后生长,骶骨前后均形成肿块;Ⅳ型,肿瘤生长仅限于骶前,骶管内无肿瘤[2]。本组8例骶骨富于细胞神经鞘瘤及6例经典型神经鞘瘤的表现均符合骶骨神经源性肿瘤的总体表现。

③④⑤⑥⑦⑧⑨⑩
图3 肿瘤细胞由梭形细胞组成,呈交织状排列 图4 肿瘤细胞具有一定的多形性 图5 肿瘤内可见核分裂象 图6 血管周围淋巴套 图7 纤维包膜增厚,散在淋巴细胞聚集 图8 肿瘤边缘漩涡样结构 图9 肿瘤组织破坏周围骨质 图10 肿瘤内灶性分布的泡沫细胞 图11 血管壁的玻璃样变 图12 肿瘤细胞S-100弥漫强阳性,EnVision法
富于细胞神经鞘瘤是由Woodruff等[3]于1981年首先报道,占所有良性神经鞘瘤的5%。临床上,富于细胞神经鞘瘤好发于脊柱旁区域,特别是纵膈、腹膜后、盆腔和骶尾部,其次为颈部和四肢。主要见于中年人,女性多见,男女比为1 ∶1.6。多表现为无痛性孤立性肿块,常在无意中发现,偶可表现为多灶性[4]。形态学上,富于细胞神经鞘瘤细胞丰富,无明显的A区和B区结构,有时细胞有异型,可见少量核分裂象,但无病理性核分裂象,也不见凝固性坏死。发生在骶骨内的病例由于送检组织破碎,包膜为部分性或局灶性,常常伴有骨质破坏。
骶骨富于细胞神经鞘瘤为少见病例,大部分文献为个案报道。国内文献共报道12例富于细胞神经鞘瘤,但发生在骶骨的仅1例[5-6]。本组8例骶骨富于细胞神经鞘瘤在患者年龄、性别及男女比上与文献报道差异均无显著性,且6例骶骨经典型神经鞘瘤也无明显差异,但发现肿瘤大小有差异。向华等[5]报道的10例肿瘤直径3~17 cm,平均7.3 cm。高金莉[6]报道的2例直径分别为10 cm、6.5 cm。本组6例对照组骶骨经典型神经鞘瘤肿块直径2.5~13 cm,平均7.2 cm。而8例骶骨富于细胞神经鞘瘤肿块直径7~24.5 cm,平均14.1 cm。从平均直径来说骶骨富于细胞神经鞘瘤要比同部位经典型神经鞘瘤和其它部位富于细胞神经鞘瘤大一倍。郭卫等[7]报道单中心原发骶骨肿瘤研究中有48例神经鞘瘤,平均最长径12 cm,孙伟等[8]曾报道一组骶骨神经鞘瘤共27例,但两组病例均未进一步进行病理分型,富于细胞神经鞘瘤的具体例数未知。从本组数据看出骶骨富于细胞神经鞘瘤约占骶骨神经鞘瘤的50%。Ki-67增殖指数平均约6%,高于经典型神经鞘瘤的3.5%,其中复发病例6.25%,初发病例5.75%。
富于细胞神经鞘瘤属于一种假肉瘤性病变[9]。需与低度恶性的恶性周围神经鞘瘤(malignant peripheral nerve sheath tumor, MPNST)相鉴别。半数以上的MPNST有Ⅰ型神经纤维瘤病,或肿瘤直接起自神经纤维瘤。大体上,MPNST与富于细胞神经鞘瘤也有所不同,MPNST多起自大神经,被覆一层假包膜,切面多呈灰褐色,常见明显坏死灶。镜下,MPNST的坏死灶呈地图状,其周围为具有异型性的分化较差的瘤细胞。缺乏富于细胞神经鞘瘤中常见的包膜下或包膜外淋巴细胞套、血管壁玻璃样变性、血管周围的淋巴细胞聚集灶和瘤细胞间的泡沫细胞等形态。此外,MPNST核分裂象较为常见,且常>10/10 HPF。免疫组化标记对两者的鉴别诊断也有帮助。S-100、CD57和GFAP标记在富于细胞神经鞘瘤为弥漫强阳性,而在大多数恶性周围神经鞘瘤中呈灶性阳性或弱阳性[10]。本组8例S-100均为强阳性,但CD57和GFAP表达不一,与文献报道略有出入。形态学上还需与纤维肉瘤、平滑肌肉瘤和滑膜肉瘤鉴别,但这三种肿瘤S-100标记均为阴性。
治疗上骶骨良性神经源性肿瘤根据肿瘤实际发病部位、侵犯骶骨和骶前情况,采用不同入路手术途径,进行边缘性切除,但由于骶骨结构的复杂性,难以保证肿瘤完全切除,所以容易复发。因此不能将富于细胞神经鞘瘤视为恶性潜能肿瘤。Casadei等[11]报道的一组70例富于细胞神经鞘瘤中有9例发生在骶骨,其中4例有复发,复发率为45%,高于平均复发率11%,未提及复发时间。本组8例富于细胞型神经鞘瘤中有4例为复发病例,复发率为50%,从第一次手术到复发的时间为3~11年,平均6.5年。而6例经典型神经鞘瘤中仅1例为复发病例,复发时间为26年。
综上,骶骨富于细胞神经鞘瘤约占骶骨神经鞘瘤的50%,相对于经典型神经鞘瘤,其体积较大、破坏周围骨质,容易复发。细胞丰富,有一定的多形性,细胞增殖指数稍高,要与MPNST相鉴别。因此,病理诊断骶骨神经鞘瘤时,如是富于细胞神经鞘瘤,应在报告中明确写出,而不能只报神经鞘瘤,这样有利于积累更多资料,供临床术后随访,及早发现肿瘤复发,避免肿瘤破坏周围骨质和软组织,也要避免过度治疗。
[1] Fletcher C D, Bridge J A, Hogendoorn P C,etal. WHO classification of tumours of soft tissue and bone[M]. Lyon: IARC Press, 2013:170-172.
[2] Guo W, Tang X, Yang Y,etal. Strategy of surgical treatment of sacral neurogenic tumors[J]. Spine, 2009,34(23):2587-2592.
[3] Woodruff J M, Godwin T A, Erlandson R A,etal. Cellular schwannoma: a variety of schwannoma sometimes mistaken for a malignant tumor[J]. Am J Surg Pathol, 1981,5(8):733-734.
[4] 王 坚, 朱雄增. 软组织肿瘤病理学[M]. 北京: 人民卫生出版社, 2008:405-407.
[5] 向 华, 王 群, 王 坚, 等. 富于细胞性神经鞘瘤的临床病理学观察[J]. 中华病理学杂志, 2005,34(4):234-235.
[6] 高金莉. 富于细胞神经鞘瘤2例临床病理分析[J]. 临床与实验病理学杂志, 2012,28(3):341-342.
[7] 郭 卫, 李大森, 蔚 然, 等. 单中心原发骶骨肿瘤790例的流行病学分析[J]. 中国脊柱脊髓杂志, 2014,24(11):971-978.
[8] 孙 伟, 马小军, 张 帆, 等. 骶骨神经源性肿瘤的外科治疗[J]. 中国骨与关节杂志, 2012,1(2):115-118.
[9] Fletcher C D, Davies S E, McKee P H. Cellular schwannoma: a distinct pseudosarcomatous entity[J]. Histopathology, 1987,11(1):21-35.
[10] Memoli V A, Brown E F, Gould V E. Glial fibrillay acidic protein(GFAP) immunoreactivity in peripheral nerve sheath tumors[J]. Ultrastuct Pathol, 1984,7(4):269-275.
[11] Casadei G P, Scheithauer B W, Hirose T,etal. Cellular schwannoma. A clinicopathologic, DNA flow cytometric, and proliferation marker study of 70 patients[J]. Cancer, 1995,75(5):1109-1119.
Sacral cellular schwannoma: a clinicopathologic analysis of eight cases
LIN Jun1, YANG Dao-hua1, HUA Ying-qi2, CHEN An3
(1DepartmentofPathology,2DepartmentofOrthopedics,3DepartmentofRadiology,ShanghaiFirstPeople’sHospital,ShanghaiJiaotongUniversitySchoolofMedicine,Shanghai200080,China)
Purpose To investigate clinicopathologic fea-tures of sacral cellular schwanoma and the difference from sacral conventional schwanoma. Methods Eight cases of sacral cellular schwanoma were collected. Microscopic examination and immunohistochemistry were performed for studying the clinical feature, radiologic appearance, pathologic characteristic, immunophenotyping, differential diagnosis and postoperative prognosis. Results There were 5 females and 3 males, whose mean age was 46.4 years. The majority of patients complained of pain in sacrococcygeal region. Radiographically, there was an endosacral or endosacral and presacal mass. Histologically, cellular schwannoma was composed of spindle cells, arranged in interlacing fascicles without nuclear palisading and Verocay bodies. Antoni A and Antoni B were not seen overtly. The destruction of bone was found. Immunohistochemically, tumor cells were diffusely and strongly positive for S-100 protein and vimentin. The mean of Ki-67 index was 6%. Tumor recurrence of 4 cases occurred several years after initial surgical resection. The mean interval to recurrence was 6.5 years. Conclusion Sacral cellular schwanoma is a rare tumor. Compared with sacral conventional schwanoma, it shows different growth pattern and pathologic features. So pathological diagnosis of the tumor should be noted for clinical follow-up and treatment.
sacrum neoplasms; cellular schwanoma; conventional schwanoma; immunohistochemistry
上海市市级医院新兴前沿技术联合攻关项目(SHDC 12013107)
上海交通大学附属第一人民医院1病理科、2骨科、3放射科 200080
林 军,男,硕士,主治医师。E-mail: forestsoldier@sohu.com
时间:2017-4-17 18:19
http://kns.cnki.net/kcms/detail/34.1073.R.20170417.1819.014.html
R 739.93
A
1001-7399(2017)04-0417-05
10.13315/j.cnki.cjcep.2017.04.014
接受日期:2016-12-02
