清金固本胶囊质量控制方法研究
2017-06-05张敏婕
李 玎,李 娜,张敏婕,陈 萍
(1.陕西中医药大学,陕西 咸阳 712046; 2.陕西省中医药研究院,陕西 西安 710003)
·检验检测·
清金固本胶囊质量控制方法研究
李 玎1,李 娜1,张敏婕1,陈 萍2
(1.陕西中医药大学,陕西 咸阳 712046; 2.陕西省中医药研究院,陕西 西安 710003)
目的 控制清金固本胶囊的质量。方法 采用薄层色谱法(TLC)对黄芩、桔梗、茯苓、防风进行鉴别,采用高效液相色谱(HPLC)法测定黄芩苷的含量。结果 黄芩苷质量浓度在4.1~32.8μg/mL范围内与峰面积线性关系良好(r=0.999 9),平均回收率为95.85%,RSD为0.71%(n=6)。结论 所建立的方法可为清金固本胶囊质量标准的建立提供参考。
清金固本胶囊;薄层色谱鉴别;高效液相色谱法;黄芩苷;含量测定;质量控制
肺癌是支气管黏膜或腺体的常见恶性肿瘤[1],其中非小细胞肺癌占肺癌的85% ~90%[2]。清金固本胶囊是陕西省中医药研究院研制的中药复方制剂,由黄芩、桔梗、茯苓、防风等10味药组方,原为临床经验方,使用多年,具有益气、清肺、化痰的功效,可用于治疗气虚、痰湿阻肺型非小细胞肺癌。本方以黄芩为君药,具有清热燥湿、泻火解毒、止血安胎功效[3],其主要活性成分为黄芩苷、黄芩素、汉黄芩素,其中黄芩苷含量最高[4]。黄芩苷能体外诱导人肺腺癌细胞株 A 549凋亡[5]。韦小白等[6]的研究表明,黄芩苷能不同程度地降低肺癌细胞增殖、迁移、侵袭的能力。桔梗能抗肺纤维化[7],茯苓、防风、党参能提高免疫力[8-10],均对肺癌的治疗有增效作用。故本研究中以黄芩苷为本品的定量指标,同时采用薄层色谱法(TLC)对方中黄芩、桔梗、茯苓、防风等进行定性鉴别。现报道如下。
1 仪器与试药
1.1 仪器
LC-1260型高效液相色谱仪,包括G1311B型四元泵,G1329B型自动进样器,G1316A型柱温箱,G4212B型二极管列检测器,TCM柱温控制器,LAB-open色谱数据工作站(美国安捷伦公司);SK2200H型超声清洗仪(昆山市超声仪器有限公司);溶剂过滤器,SYNSV0000型纯水机(美国默克密理博公司);HH-S4型恒温水浴锅(上海科伟仪器公司);101型电热鼓风恒温烘箱(北京市永光明医疗仪器厂);BP211D型十万分之一电子天平(德国赛多利斯公司)。
1.2 试药
黄芩苷对照品(批号为913-9960),对照药材黄芩(批号为 120955-200607)、桔梗(批号 121028-200608)、茯苓(批号为121117-200605)、防风(批号为120947-200405)、党参(批号为 121057-200805),均购自中国食品药品检定研究院;甲醇为色谱纯,水为超纯水,其余试剂均为分析纯;自制硅胶G、GF254板(薄层层析硅胶购自青岛海洋化工有限公司);水为超纯水;样品(规格为0.50 g/粒,批号为20150926,20151230,20160425)由陕西省中医医院制剂中心提供。
2 方法与结果
2.1 薄层色谱鉴别
2.1.1 黄芩
取样品内容物8 g,研细,加甲醇50mL,超声处理30min,滤过,滤液蒸干,残渣加水30 m L使溶解,加盐酸调pH,用乙酸乙酯提取2次,每次20 m L,合并乙酸乙酯液,蒸干,残渣加甲醇1m L使溶解,即得供试品溶液;称取黄芩对照药材1 g,同法制成黄芩对照药材溶液;取黄芩苷对照品适量,加甲醇制成1 mL含1 mg的溶液,作为对照品溶液;按处方比例称取除黄芩外的其他药材适量,制成缺黄芩的阴性样品,按供试品溶液制备方法制成缺黄芩的阴性对照品溶液。
按 2015年版《中国药典(一部)》方法,照薄层色谱法(通则 0502)试验,取上述各溶液 10μL,分别点于同一硅胶G薄层板上,以乙酸乙酯-丁酮-甲酸-水(5∶3∶1∶1)为展开剂,预饱和30min,展开,取出,晾干,喷以2%三氯化铁乙醇溶液。
供试品溶液色谱中,在与对照药材溶液色谱相应位置上,显相同颜色的荧光斑点;在与对照品溶液色谱相应位置上,显相同颜色的荧光斑点,阴性对照无干扰。详见图1。
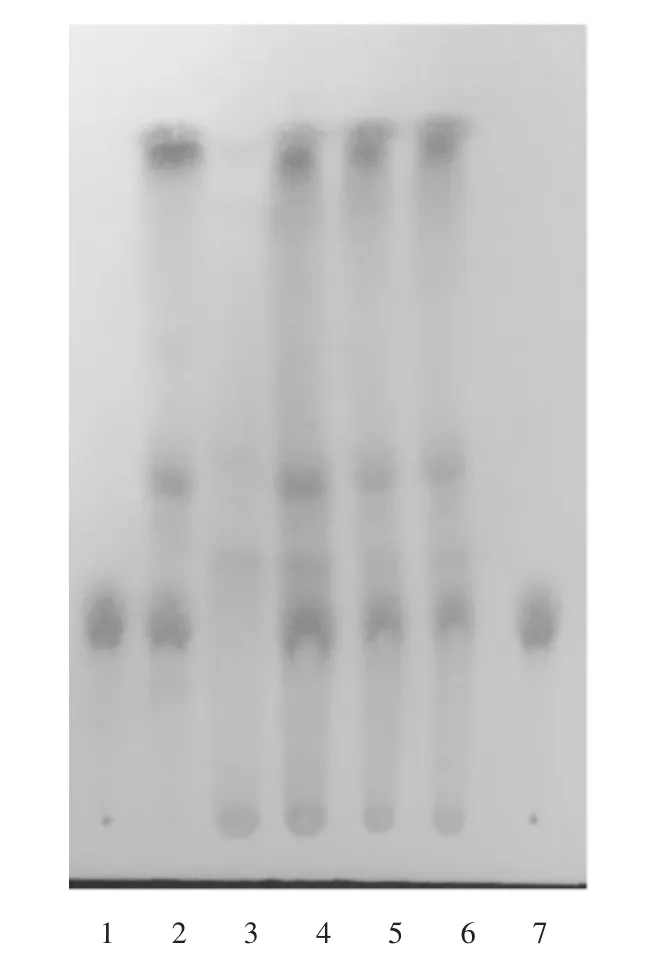
1,7.黄芩苷对照品溶液 2.黄芩对照药材溶液3.阴性对照品溶液 4-6.供试品溶液图1 黄芩薄层色谱图
2.1.2 桔梗
取样品内容物8 g,研细,加7%硫酸乙醇-水(1∶3)混合溶液20mL,加热回流3 h,放冷,用三氯甲烷提取2次,每次20m L,合并三氯甲烷液,用水洗涤2次,每次30mL,弃去洗液,三氯甲烷液用无水硫酸钠脱水,滤过,滤液蒸干,残渣加甲醇1 m L使溶解,得供试品溶液;取桔梗对照药材1 g,同法制成桔梗对照药材溶液;按处方比例称取除桔梗外的其他药材,制成阴性对照品,按供试品溶液制备方法制成缺桔梗的阴性对照品溶液。
按2015年版《中国药典(一部)》方法,照薄层色谱法(通则0502)试验,取上述各溶液10μL,分别点于同一硅胶G254薄层板上,以三氯甲烷-乙醚(2∶1)为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液。
供试品溶液色谱中,在与对照药材溶液色谱相应位置上,显相同颜色的荧光斑点,阴性对照无干扰,故该方法纳入质量标准中。详见图2。
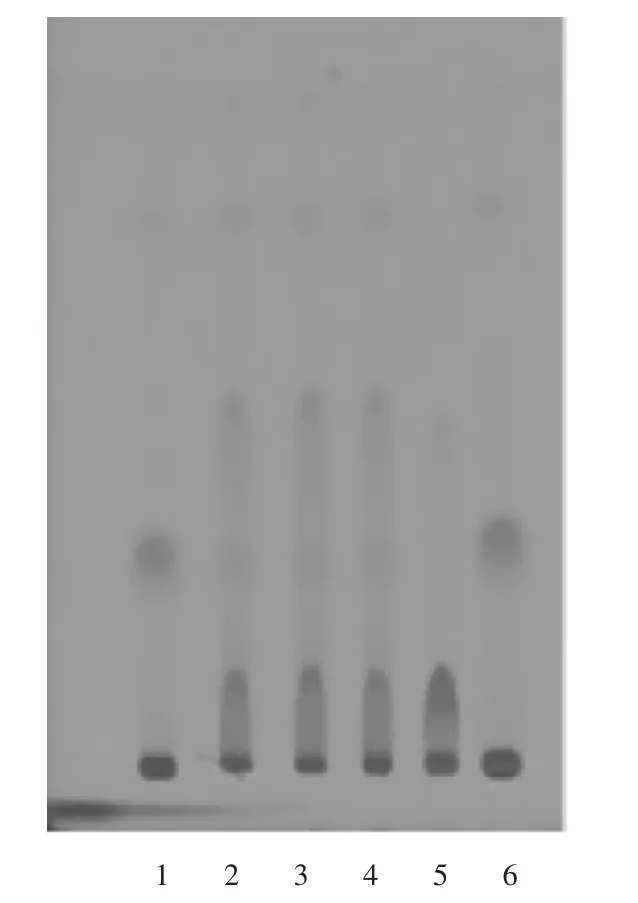
1,6.桔梗对照药材溶液 2-4.供试品溶液5.阴性对照品溶液图2 桔梗薄层色谱图
2.1.3 茯苓
取样品内容物12 g,研细,加乙醚60m L,超声处理30min,滤过,滤液挥干,残渣加无水乙醇1m L使溶解,即得供试品溶液;取茯苓对照药材1 g,同法制成茯苓对照药材溶液,按处方比例称取除茯苓外的其他药材适量,制成缺茯苓的阴性样品,按供试品溶液制备方法制成缺茯苓的阴性对照品溶液。
按2015年版《中国药典(一部)》方法,照薄层色谱法(通则0502)试验,取上述各溶液20μL,分别点于同一硅胶G薄层板上,以乙醚-石油醚(1∶1,30~60℃)为展开剂,预饱和30min,展开,取出,晾干,置紫外光灯(365 nm)下检视。
供试品溶液色谱中,在与茯苓对照药材溶液色谱相应位置上,显相同颜色的荧光斑点,阴性对照无干扰,故该方法纳入质量标准中。详见图3。
2.1.4 防风
取样品内容物5 g,加水100mL,用水饱和正丁醇15 m L提取2次,合并正丁醇液,用0.1 mol/L氢氧化钠溶液洗涤2次,每次20 m L,弃去碱液,蒸干正丁醇液,残渣加甲醇1mL使溶解,即得供试品溶液;取防风对照药材1 g,同法制成防风对照药材溶液;按处方比例称取除防风外的其他药材适量,制成缺防风的阴性样品,按供试品溶液制备方法制成缺防风的阴性对照品溶液。
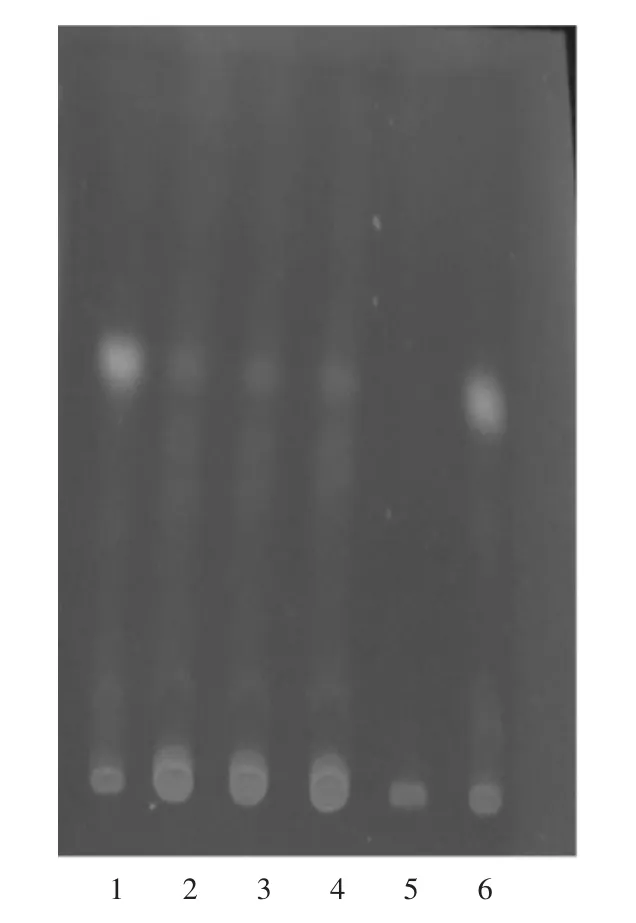
1,6.茯苓对照药材溶液 2-4.供试品溶液5.阴性对照品溶液图3 茯苓薄层色谱图
按2015年版《中国药典(一部)》,照薄层色谱法(通则0502)试验,取上述各溶液10μL,分别点于同一硅胶G薄层板上,以三氯甲烷-乙酸乙酯-甲醇-甲酸(12∶4∶3∶0.2)为展开剂,预饱和30min,展开,取出,晾干,喷以10%硫酸乙醇溶液,置紫外光灯(365 nm)下检视。
供试品溶液色谱中,在与防风对照药材溶液色谱相应位置上,显相同颜色的荧光斑点,阴性对照无干扰,故该方法纳入质量标准中。详见图4。
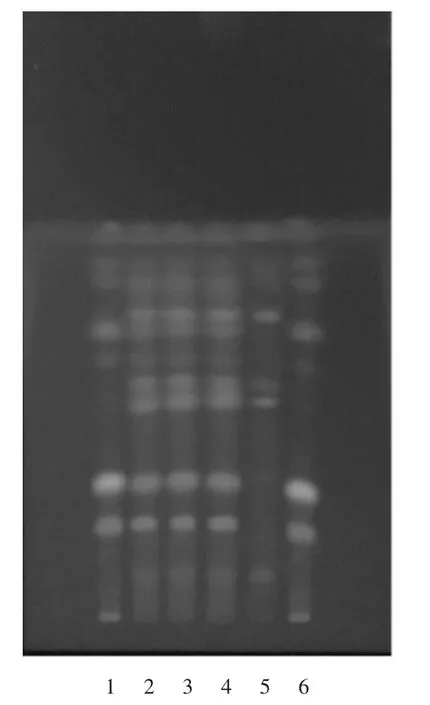
1,6.防风对照药材溶液 2-4.供试品溶液5.阴性对照品溶液图4 防风薄层色谱图
2.1.5 党参
取样品内容物6 g,加甲醇30m L,回流提取1.5 h,滤过,滤液挥干,残渣加丙酮1m L使溶解,即得供试品溶液;取党参对照药材1 g,同法制成党参对照药材溶液;按处方比例称取除党参外的其他药材适量,制成缺党参的阴性样品,按供试品溶液制备方法制成缺党参的阴性对照品溶液。
按2015年版《中国药典(一部)》,照薄层色谱法(通则0502)试验,取上述各溶液10μL,分别点于同一硅胶G薄层板上,以三氯甲烷-甲醇-冰醋酸-水(15∶10∶1∶0.5)为展开剂,预饱和30 min,展开,取出,晾干,喷以磷钼酸乙醇溶液,于105℃加热显色。
供试品溶液色谱中,在与党参对照药材溶液色谱相应位置上,显相同颜色的荧光斑点,阴性对照无干扰,故该方法纳入质量标准中。详见图5。

1.党参对照药材溶液 2-4.供试品溶液5.阴性对照品溶液图5 党参薄层色谱图
2.2 黄芩苷含量测定
2.2.1 色谱条件与系统适用性试验
色谱柱:KromasilC18不锈钢色谱柱(250mm×4.6mm,5μm);流动相:甲醇-水-冰醋酸(46∶54∶1);流速:1.0 mL/min;柱温:(30.0±0.5)℃。经紫外光谱于190~400 nm波长范围内扫描,结果黄芩苷在276 nm波长处有最大吸收,故确定检测波长为276 nm。在此色谱条件下,测得供试品溶液中黄芩苷色谱峰可以基本达到基线分离,黄芩苷色谱峰理论板数(N)大于2 000,拖尾因子(T)为1.01,与相邻峰的分离度大于1.5。
2.2.2 溶液制备
精密称取黄芩苷对照品,置 50 mL容量瓶中,加70%甲醇溶解,定容至刻度,即得对照品溶液;取本品内容物 1.0 g,精密称定,置 100 mL具塞三角瓶中,加70%甲醇适量,加热回流,提取2次,每次1 h,取出,滤过,合并滤液,加70%甲醇定容于100m L容量瓶中,摇匀,用微孔滤膜(0.45μm)滤过,即得供试品溶液。
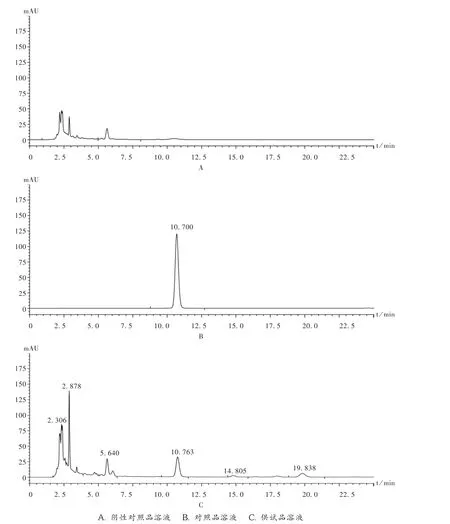
图6 黄芩苷高效液相色谱图
2.2.3 方法学考察
阴性干扰试验:按处方比例,照本品制备工艺,制成不含黄芩的阴性对照品样品,再按供试品溶液制备方法制成阴性对照品溶液。精密吸取5μL,注入液相色谱仪。结果其色谱图在黄芩苷峰位置上无色谱峰出现,阴性对照无干扰。详见图6。
线性关系考察:精密称取黄芩苷对照品,加70%甲醇制成质量浓度为0.082 g/L的溶液,作为对照品溶液。精密吸取对照品溶液0.5,1.0,2.0,3.0,4.0,5.0m L,加70%甲醇定溶于10 m L容量瓶中,均进样10μL,按拟订色谱条件进样测定。以峰面积(Y)为纵坐标、质量浓度(X)为横坐标进行线性回归,得回归方程 Y=1.981 878X+1.198 434,r=0.996 7(n=6)。结果表明,黄芩苷质量浓度在 4.1~41.0μg/mL范围内与峰面积线性关系良好。
精密度试验:分别吸取上述对照品溶液与供试品溶液各10μL,分别重复进样5次。结果对照品溶液色谱峰面积的 RSD为0.48%(n=6),供试品溶液色谱峰面积的 RSD为0.66%(n=6),表明仪器精密度良好。
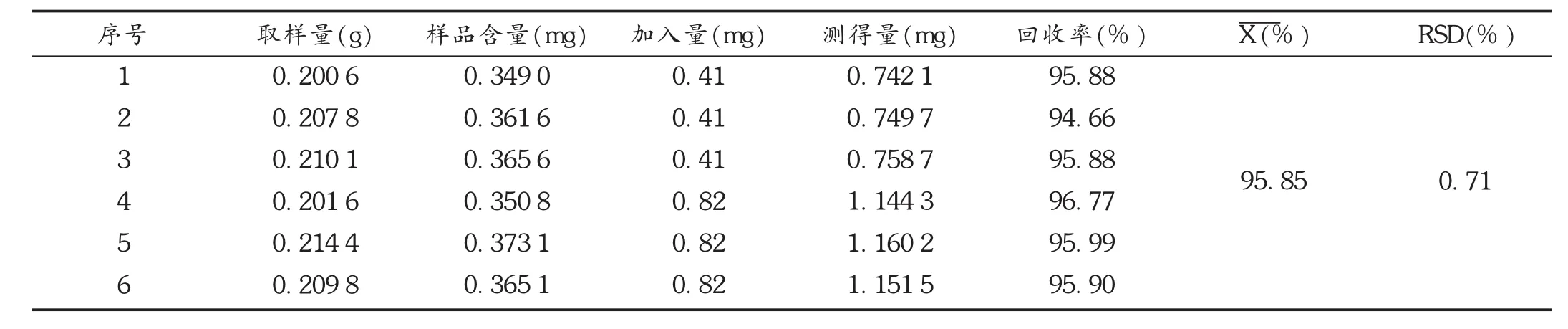
表1 黄芩苷加样回收试验结果(n=6)

表2 样品含量测定结果(mg/粒,n=2)
稳定性试验:取同一份供试品溶液,分别于0,1,3,6,12,24 h时按拟订色谱条件进样测定。结果黄芩苷峰面积的 RSD为1.59%(n=6),供试品溶液在6 h以前稳定性良好,10 h和24 h峰面积的变化无规律性,表明供试品溶液在12 h内稳定性较好。
重复性试验:取同一批样品6份,按拟订方法进样测定,并计算含量。结果平均含量为每粒0.87mg,RSD为3.63%(n=6),表明方法重复性较好。
加样回收试验:取已知含量的样品粉末约200mg,共6份,精密称定,分别置100m L容量瓶中,分别精密加入黄芩苷对照品溶液(0.82 g/L)0.5,1.0m L,按拟订色谱条件进样测定,并计算回收率。结果平均回收率为95.84%,RSD为0.7%(n=6)。详见表1。
2.2.4 样品含量测定及限度确定
取3批样品各2份,依法制备供试品溶液,并进样测定,结果见表2。根据3批样品测定结果,暂定本品每粒含黄芩以黄芩苷(C21H18O11)计不得少于0.90mg。
3 讨论
本研究中对方中枳壳、陈皮、白术、夏枯草进行了薄层色谱鉴别,但阴性样品存在干扰,暂不能纳入标准,故有待进一步研究。
有关黄芩苷的测定,依据黄芩苷理化性质及查阅文献[11-15],本研究中分别采用热回流、超声方法。结果表明,回流比超声测得黄芩苷含量更高,确定以70%甲醇回流提取60min,提取率最高,且其他组分干扰小。
[1]张亚琨,郭其森.非小细胞肺癌的化疗进展[J].中国医药科学,2011,15(1):40-42.
[2]Chang JTH,Lee YM,Huang RS.The impactof the Cancer Genome Atlas on lung cancer[J].Translational Research,2015,166(6):568-585.
[3]国家药典委员会.中华人民共和国药典(一部)[M].北京:中国医药科技出版社,2015:301.
[4]王宏志,喻春皓,高 钧,等.HPLC分析比较炮制和提取方法对黄芩活性成分的影响[J].中国中药杂志,2007,32(16):1637.
[5]郑海峰,王英妹,武铁军,等.黄芩苷对人肺腺癌A549细胞的抑制作用及相关机制[J].中成药,2012,34(11):2214-2216.
[6]韦小白,董竞成.黄芩苷对人肺腺癌LTEP-A2细胞的抑制作用及机制研究[J].世界中医药,2014,9(2):213-217.
[7]于维颖,祝红杰.桔梗治疗支气管哮喘的药理机制研究[J].中医药学报,2012,40(3):38-40.
[8]刘媛媛,陈友香,侯安继.羟甲基茯苓多糖对小鼠 T淋巴细胞分泌细胞因子的影响[J].中药药理与临床,2006,22(3/4):71-72.
[9]王 玲.防风对免度功能的影响[J].实用医技,1999,6(2):139.
[10]张华荣,姜国辉.党参的药理与临床研究进展[J].中医药信息,1996(5):17-21.
[11]张冬梅,桑 媛,刘 冰.HPLC法同时测定双黄连注射液中绿原酸及黄芩苷含量[J].药物分析杂志,2005,25(10):1279-1280.
[12]郭 忠.双黄连制剂含量分析方法综述[J].中国药事,2004,18(3):194-196.
[13]王 猛,高景会.HPLC法测定清热解毒片中黄芩苷的含量[J].黑龙江医药,2015,28(2):219-221.
[14]陈叶青,陈凡凡,金佩芬,等.HPLC法测定通窍耳聋丸中橙皮苷和黄芩苷的含量[J].解放军药学学报,2015,31(4):324-326.
[15]张淑瑜,于燕莉,毕云生,等.退烧颗粒中黄芩苷的成分鉴别和含量测定[J].解放军药学学报,2015,31(1):65-67.
Quality Control of Qingjinguben Capsule
Li Ding1,Li Na1,Zhang Minjie1,Chen Ping2
(1.Shaanxi University of Chinese Medicine,Xianyang,Shaanxi,China 712046; 2.Shaanxi Province Chinese Medicine Research Institute,Xi′an,Shaanxi,China 710003)
Ob jective To control the quality of Qingjinguben Capsule.M ethods TLC method was used to identify Scutellariae Radix,Platycodonis Radix,Poria,Saposhnikoviae Radix,Codonopsis.The content of baicalin was determined by HPLC.Results The chromatographic spots were clear with good separation,the chromatogram for identification by TLC was distinct and highly specific.The linear range was 4.1-32.8μg/m l(r=0.999 9),the average recovery rate was 95.85%,RSD was 0.71%(n=6).Conclusion The method can provide reference for the establishment of quality standard of Qingjinguben Capsule.
Qingjinguben Capsule;TLC;HPLC;baicalin;content determination;quality contorl
R284.1
A
1006-4931(2017)08-0012-05
2016-12-08;
2017-01-22)
10.3969/j.issn.1006-4931.2017.08.004
陕西省自然科学基础研究计划项目[2 0 1 2 JM4 0 5 3]。
李玎(1992-),女,在读硕士研究生,研究方向为中药制剂工艺及质量标准,(电子信箱)lidingle19@163.com。
陈萍(1 9 6 4-),女,研究员,硕士研究生导师,主要从事中药化学与中药新药研究,(电子信箱)c p 3 0 4 9 0 3 3@1 6 3.c om。
