X连锁遗传的Emery-Dreifuss肌营养不良一家系的临床、病理及基因突变特点
2017-06-01赵丹华赵运涛杨旭温宏峰徐依成王培福李继来杜继臣
赵丹华 赵运涛 杨旭 温宏峰 徐依成 王培福 李继来 杜继臣
X连锁遗传的Emery-Dreifuss肌营养不良一家系的临床、病理及基因突变特点
赵丹华 赵运涛 杨旭 温宏峰 徐依成 王培福 李继来 杜继臣
目的 报道1例X连锁遗传的Emery-Dreifuss肌营养不良(X-EDMD)患者家系的临床、病理及基因突变特点。方法 总结作者单位收治的1例X-EDMD患者的临床及骨骼肌病理特点,并进行EMD及LMNA基因突变检测,同时对患者家系谱进行分析。结果 该家系中有4例患者,主要表现为缓慢型心律失常,2例已猝死,先证者同时有轻度肘关节挛缩及肌萎缩。先证者心电图检查示房性心动过速合并Ⅲ度房室阻滞,交界区逸搏心律;超声心动图检查示全心扩大,二尖瓣及三尖瓣A峰消失,射血分数60%。肌电图检查示右肱二头肌、左第一骨间肌、左腓肠肌、左股四头肌运动单位电压均增高,神经传导未见异常。左肱二头肌活检示肌营养不良样改变,免疫组化染色显示肌核无伊默菌素蛋白表达。基因分析显示先证者存在EMD基因插入突变c.650_654dupTGGGC,为已报道的致病性突变,LMNA基因均未发现致病性突变。结论EMD基因c.650_654dup TGGGC插入突变导致的X-EDMD患者心脏损害突出,以心房的电活动-机械活动及结构的严重受累为特征,而肌萎缩及关节挛缩症状相对轻微。
基因,X连锁;肌营养不良,Emery-Dreifuss型;EMD基因
Emery-Dreifuss肌营养不良(Emery-Dreifuss muscular dystrophy,EDMD)是一种相对罕见的肌营养不良,于1962年首次报道,1966年被Emery及Dreifuss确认为一种独立的疾病[1]。本病于儿童早期发病,临床特征为早期出现的关节挛缩、缓慢进展的肌无力和肌萎缩以及心脏传导异常三联征[2]。EDMD具有X连锁遗传(X-EDMD)、常染色体显性(autosomal dominant,AD)或隐性遗传(autosomal recessive,AR)3种不同的遗传方式,致病基因具有明显的异质性。EMD及FHL1基因突变可导致X-EDMD,而LMNA、Nesprin-1、Nesprin-2及TMEM43基因突变则可以导致AD-EDMD和(或)AR-EDMD,另有50%~60%的患者致病基因不明[3-5]。EMD基因定位于Xq28,包含2100个碱基,编码产物为伊默菌素(emerin),该蛋白锚定于骨骼肌、心肌和平滑肌的核内膜上。目前已知EMD基因致病突变超过200个,分布于整个基因,而国内目前只报道了1个缺失突变(c.26_39delATACCGAGCTGACC)[6]。本文报道另一个已知EMD基因插入突变导致的X-EDMD家系,旨在探讨该家系的临床表型及骨骼肌病理改变特点。
1 家系报告
先证者男性,38岁,主因“间断乏力半年”于2015-08-12就诊于航天中心医院心内科。患者半年前无明显诱因间断出现一过性乏力,无胸痛、胸闷,无头晕及双眼黑矇,无肢体力弱及肌肉萎缩,伴体重进行性下降10 kg,于当地医院查心电图结果显示:Ⅲ度房室阻滞,交界区逸搏心律(心率40次/min),为求进一步诊治遂至作者医院就诊。患者足月顺产,出生时无异常,满月后即被家人发现双侧肘关节轻度屈曲,未诊治,自幼智力及运动发育均无异常,平时活动耐力较同龄人稍差,但不影响日常运动。家族中外公(Ⅰ-1)年轻时猝死,原因不详;大舅(Ⅱ-1)74岁时诉“心脏不好”,具体不详;大姨(Ⅱ-2)50岁时发现心率慢,当地医院建议行心脏起搏器治疗,但未予采纳,后猝死,其长子(Ⅲ-4)10余岁时发现心率慢,未治疗,现45岁。患者母亲、另1位姨妈、另3位舅舅及其余同辈表兄妹均无明确心脏疾病史。除先证者外,其余家族成员均否认肌无力、肌肉萎缩及关节挛缩史(家系图谱见图1)。入院查体:双肘关节轻度挛缩。心尖搏动位于第5肋间左锁骨中线外1 cm,心率40次/min,心律齐,心音有力。实验室检查:血肌酸激酶(CK)308.0 IU/L,CK-MB同工酶20.3 IU/L;心电图检查示房性心动过速合并Ⅲ度房室阻滞,交界区逸搏心律(心率38次/min),因P波振幅极低,故进一步行食道心电图检查证实患者存在房性心动过速;心脏彩超检查显示,静息状态下全心扩大,二尖瓣、三尖瓣及肺动脉瓣少量反流,二尖瓣及三尖瓣A峰消失,肺动脉收缩压43 mmHg(1 mmHg=0.133 kPa),射血分数60%;心脏MRI检查结果显示,左、右心房增大,左心室增大;心肌灌注成像显示心肌各节段未见明确灌注缺损及延迟;心肌活性扫描显示心肌各节段未见明确延迟强化影。于心内科行心脏起搏器植入术,术后转入神经内科进一步行病因诊断。神经系统查体:意识清楚,言语流利,高级皮层功能及脑神经检查无异常,四肢肌力5级,双侧冈上肌、冈下肌轻度萎缩,双侧腓肠肌略肥大,四肢腱反射对称引出,病理征阴性。针极肌电图检查示:右肱二头肌、左第一骨间肌、左腓肠肌、左股四头肌运动单位电压均增高,所检神经传导未见异常。经患者知情同意,行左肱二头肌活检,骨骼肌病理结果示:肌束衣内结缔组织轻度增生,可见散在分布的部分小圆状及个别小角状萎缩肌纤维,部分肌纤维肥大变圆,可见个别新鲜坏死及再生肌纤维,伴随明显的肌纤维分裂及核内移现象,肌纤维内肌核数目明显增多;ATP酶染色显示两型肌纤维在个别区域呈小片状分布,肥大及萎缩肌纤维均累及两型;免疫组织化学染色显示Dystrophin-N、C、R,Dysferlin及α、β、γ-Sarcoglycan染色均正常,Desmin染色可见萎缩肌纤维深染,肌核上无Emerin表达(图2)。进一步行基因检测显示,先证者EMD基因存在有害插入突变c.650_654dupTGGGC(图3),为已报道的致病性突变,导致正常的p.219-p.254氨基酸缺失,被19个异常的氨基酸取代;LMNA基因未见致病性突变。

图1 X-EDMD患者的家系谱图
2 讨论
本文先证者为中年男性,主要表现为房性心动过速合并Ⅲ度房室阻滞,交界区逸搏心律,伴轻度的肌萎缩及肘关节挛缩,CK轻度升高,左肱二头肌活检病理检查示肌营养不良样改变,Emerin蛋白在肌核上无表达,EMD基因检测证实存在致病性插入突变c.650_654dupTGGGC,故诊断为EDMD。家系中先证者、先证者的外公及表哥均发病,而先证者的母亲临床表型正常,符合X-连锁遗传的特点。先证者的大姨为女性携带者,也出现了心脏损害。这与文献报道一致,故女性携带者应定期进行心电图及心脏超声的检查[7],以便及早进行干预治疗。

注:A:HE染色示肌束衣内结缔组织轻度增生,肌纤维直径变异加大,可见肌纤维分裂及核内移;B:ATP酶染色示I型肌纤维呈小片状分布(pH 4.4);C:ATP酶染色示Ⅱ型肌纤维呈小片状分布(pH 10.7);D:NADH染色显示坏死及分裂肌纤维;E:Emerin染色显示肌核无着色;F:Emerin染色显示肌核深染(阳性对照) 图2 先证者左肱二头肌病理检查结果
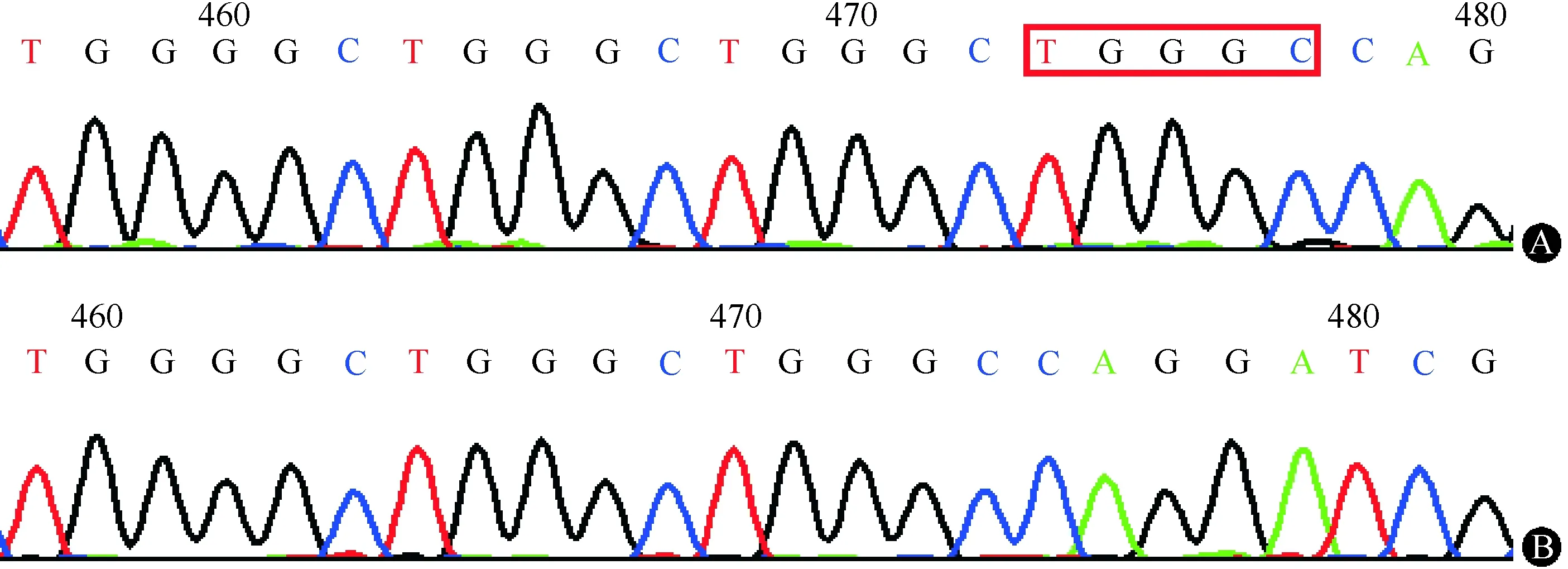
注:A:PCR产物直接测序显示先证者EMD基因存在c.650_654dupTGGGC插入突变;B:正常对照 图3 先证者EMD基因测序分析结果
经典的EDMD的临床三联征包括:(1)早于肌无力出现的关节挛缩,包括肘关节、跟腱及颈后伸肌,后期可发展至整个脊柱活动受限;(2)缓慢进展的肌无力及肌萎缩,早期呈特征性的肱腓型分布,后期发展成肢带型分布,但肌无力相对较轻;(3)心脏传导异常,包括窦性心动过缓、PR间期延长及房室阻滞,严重时心脏骤停,需植入心脏起搏器,且可并发扩张型心肌病[8]。本家系先证者的临床表现符合X-EDMD的临床特点,但与典型临床表现相比又存在以下不同:首先,患者虽有轻度的肘关节挛缩,但没有出现跟腱挛缩及脊柱强直,这与文献中报道的轻微关节挛缩相似[9];其次,患者仅有肩带肌轻度萎缩,而四肢肌力正常,不符合经典的肱腓型分布,但EMD基因突变导致的肢带型肌无力及肌萎缩其实并不少见,而患者出现双侧腓肠肌肥大,这一特点更常见于LMNA基因突变[7];第三,先证者的心脏损害十分突出,存在房性心动过速,提示心房电活动异常,双心房扩大提示心房结构异常,超声心动图示二/三尖瓣无A峰,提示双房机械静止,故心房在电活动、结构及机械活动3个层面上均发生了严重损害,其受累严重程度远大于心室。心房静止合并房室阻滞可见于X-EDMD患者,本文先证者虽然无心房机械活动,但仍有心房电活动。由于房性心动过速合并Ⅲ度房室阻滞非常罕见,因此这一表现也许是X-EDMD患者心脏受累的特征性改变。
EDMD患者的骨骼肌病理以肌营养不良样改变为主,核内移及核链常见,且肌纤维内肌核的数目明显增多,伴随核肥大、核萎缩及核周空泡[10]。电镜下可见核内染色质聚集,染色质及核膜之间的连接断裂。本文先证者的病理改变除有上述特点以外,还可见明显的肌纤维分裂及小的肌源性群组化,而其肌电图显示不同部位、多块肌肉的运动单位电位波幅升高,提示肌纤维密度增加,推测可能是肌纤维分裂所致[11],并非只见于神经源性损害,故先证者的电生理改变与其病理改变一致。
目前研究认为2/3的X-EDMD患者为EMD基因突变所致[12]。EMD基因编码254个氨基酸,前220个氨基酸构成蛋白的氨基端,位于核内,最后23个氨基酸组成羧基端,为一段跨膜蛋白,位于核内膜上。刚合成的Emerin嵌入内质网中,进行转录后修饰,然后弥散到至核内,通过羧基端与核纤层蛋白结合,其主要功能是在肌肉收缩过程中对抗机械性压力以稳定核膜。本文先证者EMD基因存在插入突变c.650_654dupTGGGC,导致正常的p.219-p.254氨基酸缺失,被19个异常的氨基酸取代,由此整个羧基端的长度和构象均发生改变,其疏水性明显降低,由此可能引起Emerin蛋白被降解或是被错误定位在胞浆中而致病[13]。
除本文报道的家系外,目前共报道了2例EMD基因c.650_654dupTGGGC插入突变所致的散发X-EDMD患者。1例为17岁男性,表现为肘部及颈部关节挛缩、肢带型肌无力以及房室阻滞[14],另1例患者临床表型不详[7]。由于病例数较少,该基因突变所致的临床特点尚不明确。但有文献报道,EMD基因突变所致的临床表型主要为心脏受累,而肌无力及关节挛缩可以相对较轻[7]。本家系患者即符合这一特点。需注意的是,本文先证者的心脏损害以心房电活动-机械活动及结构的严重受累为特征,表现为罕见的房性心动过速合并Ⅲ度房室阻滞,这一特殊的临床表型是否是X-EDMD患者的特征性表现有待进一步探讨。
志谢:感谢北京大学第一医院神经病理室张秋荣、左越换及刘婧技师给予的病理技术支持。
[1]Emery AE,Dreifuss FE. Unusual type of benign X-linked muscular dystrophy[J]. J Neurol Neurosurg Psychiat,1966,29(4):338-342.
[2]Emery AE. Emery-Dreifuss muscular dystrophy—40 year retrospective[J]. Neuromuscul Disord,2000,10(4-5):228-232.
[3]Gueneau L, Bertrand AT,Jais JP.Mutations of the FHL1 gene cause Emery-Dreifuss muscular dystrophy[J]. Am J Hum Genet,2009,85(3):338-353.
[4]Zhang Q,Bethmann C,Worth NF,et al. Nesprin-1 and -2 are involved in the pathogenesis of Emery Dreifuss muscular dystrophy and are critical for nuclear envelope integrity[J]. Hum Mol Genet,2007,16(23):2816-2833.
[5]Liang WC,Mitsuhashi H,Keduka E,et al. TMEM43 mutations in Emery-Dreifuss muscular dystrophy-related myopathy[J]. Ann Neurol,2010,69(6):1005-1013.
[6]Zhang M,Chen J,Si D,et al. Whole exome sequencing identifies a novel EMD mutation in a Chinese family with dilated cardiomyopathy[J]. BMC Med Genet,2014,15:77.
[7]Astejada MN,Goto K,Nagano A,et al. Emerinopathy and laminopathy clinical, pathological and molecular features of muscular dystrophy with nuclear envelopathy in Japan[J]. Acta Myol,2007,26(3):159-164.
[8]Helbling-Leclerc A,Bonne G,Schwartz K. Emery-Dreifuss muscular dystrophy[J]. Eur J Hum Genet,2002,10(3):157-161.
[9]Ura S,Hayashi YK,Goto K,et al. Limb-girdle muscular dystrophy due to emerin gene mutations[J]. Arch Neurol,2007,64(7):1038-1041.
[10]Menezes MP,Waddell LB,Evesson FJ,et al. Importance and challenge of making an early diagnosis in LMNA-related muscular dystrophy[J]. Neurology,2012,78(16):1258-1263.
[11]汤晓芙. 临床肌电图学[M].北京:北京医科大学中国协和医科大学联合出版社,1995.100-113.
[12]Pillers DA,Von Bergen NH. Emery-Dreifuss muscular dystrophy:a test case for precision medicine[J]. Appl Clin Genet,2016,9:27-32.
[13]Méjat A,Misteli T. LINC complexes in health and disease[J]. Nucleus,2010,1(1):40-52.
[14]Klauck SM,Wilgenbus P,Yates JR,et al. Identification of novel mutations in three families with Emery-Dreifuss muscular dystrophy[J]. Hum Mol Genet,1995,4(10):1853-1857.
(本文编辑:时秋宽)
The clinical, myopathological and genetic features of a family with X-linked Emery-Dreifuss muscular dystrophy
ZHAODanhua,ZHAOYuntao,YANGXu,WENHongfeng,XUYicheng,WANGPeifu,LIJilai,DUJichen*.
#Department of Neurology, Aerospace Center Hospital,Aerospace Clinical Medical College Affiliated to Peking University, Beijing 100049, China
Corresponding author:DU Jichen, Email:djc189@tom.com
Objective To report the clinical,pathological and genetic features of a Chinese family with X-linked Emery-Dreifuss muscular dystrophy (X-EDMD). Methods The clinical and muscular pathological features of a family with X-EDMD were summarized. The sequence ofEMDandLMNAgene was analyzed after genomic DNA was extracted from the white blood cells of the proband. Results The four affected members in the family mainly manifested as bradycardia, two of which underwent sudden cardiac death. The proband also had mild contracture of elbow and muscular atrophy. Electrocardiogram showed atrial tachycardia accompanied by third-degree atrioventricular block with a junctional escape rhythm. Echocardiography revealed enlargement of the whole heart and lack of “A” wave in the Doppler mitral/ tricuspid flow pattern, with an ejection fraction of 60%. Electromyography disclosed high potential of motor unit in right biceps, left first dorsal interosseous, left gastrocnemius muscles and left quadriceps, with normal nerve conduction velocities. The biopsy of left biceps disclosed muscular dystrophy. Immunohistochemistry showed deficient expression of emerin in all nuclei of muscle fibers. Genetic analysis revealed a pathogenic insertion mutation, c.650_654dupTGGGC, inEMDgene in the proband, and no mutation inLMNAgene. Conclusions The insertion mutation of c.650_654dupTGGGC inEMDgene resulted in a family with X-EDMD. The affected members suffered from severe cardiac involvement which was characterized by serious damage of atrial electrical activity, mechanical activity and structure, as well as mild muscular atrophy and joint contracture.
genes, X-linked; muscular dystrophy, Emery-Dreifuss;EMDgene
10.3969/j.issn.1006-2963.2017.03.011
100049北京大学航天临床医学院 航天中心医院神经内科(赵丹华、杨旭、温宏峰、徐依成、王培福、李继来、杜继臣),心内科(赵运涛)
杜继臣,Email:djc189@tom.com
R746.4
A
1006-2963(2017)03-0197-04
2016-10-02)
