无水三氯化铝催化的吲哚自身偶联反应研究
2017-05-25于静文宋璐娜
于静文,宋璐娜
(山西医科大学 晋祠学院 基础医学部,山西 太原 030025)
无水三氯化铝催化的吲哚自身偶联反应研究
于静文,宋璐娜
(山西医科大学 晋祠学院 基础医学部,山西 太原 030025)
提出了一种由无水三氯化铝代替质子酸或酸酐作催化剂实现吲哚自身偶联反应的新方法。该方法使用了廉价的催化剂、操作简便、高收率(86%-90%)且反应时间短(小于40 min)。此反应的原子利用率为100%,在有机合成中具有一定的应用。通过核磁共振氢谱、核磁共振碳谱对产物的结构进行了表征。另外文中还提出了可能的反应机理。
合成方法;吲哚;无水三氯化铝;自身偶联
0 引 言
吲哚(2,3-苯并吡咯)是一种重要的含氮杂环化合物,它是有机合成的重要中间体,在医药、农药、香料、食品、饲料添加剂和染料等方面具有广泛的应用[1]。吲哚由于其独特的生物活性和药理作用[2],使它已成为生物、医药和材料交叉领域的明星分子[3],多年来一直吸引着化学工作者对其结构进行修饰和进行仿生全合成研究[4-5]。自然界含吲哚环系的化合物分布很广,自1983年以来已经发现了3 500多种,其中许多都具有重要的生物活性,与生命活动密切相关[6]。对吲哚自身而言,化学反应也极其丰富,可通过各种方法进行衍生化[7]。在吲哚分子结构中,C-3位的反应活性最强[8],与其相关的反应类型最多,因此在众多的衍生物中C-3位取代的吲哚最引人瞩目[9]。
相关文献报道,吲哚在质子酸或酸酐的催化作用下,会发生一种自身偶联反应,这种反应的原子利用率为100%,具有十分重要的研究价值。2011年,Norio Shibata课题组报道了吲哚在三氟乙酸酐的催化作用下搅拌过夜可发生二聚生成吲哚偶联产物[10];2013年,席真等人报道了吲哚在饱和氯化氢气体的乙醚溶液中搅拌18 h后也可得到同样的产物[11]。虽然两种方法都成功地实现了吲哚的自身偶联,但仍存在一定缺陷:(1)反应时间较长(例如18 h或过夜);(2)产物收率较低(例如63%);(3)准备工作烦琐(例如需制备饱和氯化氢气体的乙醚溶液)。因此,探寻一种适宜的反应条件实现吲哚的自身偶联仍是一个亟待解决的问题。
众所周知,无水三氯化铝是一种经济易得、便于操作、使用较为广泛的路易斯酸催化剂[12]。本文尝试使用无水三氯化铝代替氯化氢气体和三氟乙酸酐催化吲哚的自身偶联反应。实验表明,在无水三氯化铝作催化剂,0℃和二氯甲烷作溶剂的条件下,反应在40 min内即可完成,本文最终实现了三种吲哚自身偶联产物的合成,收率可达86%-90%。
反应方程式为:
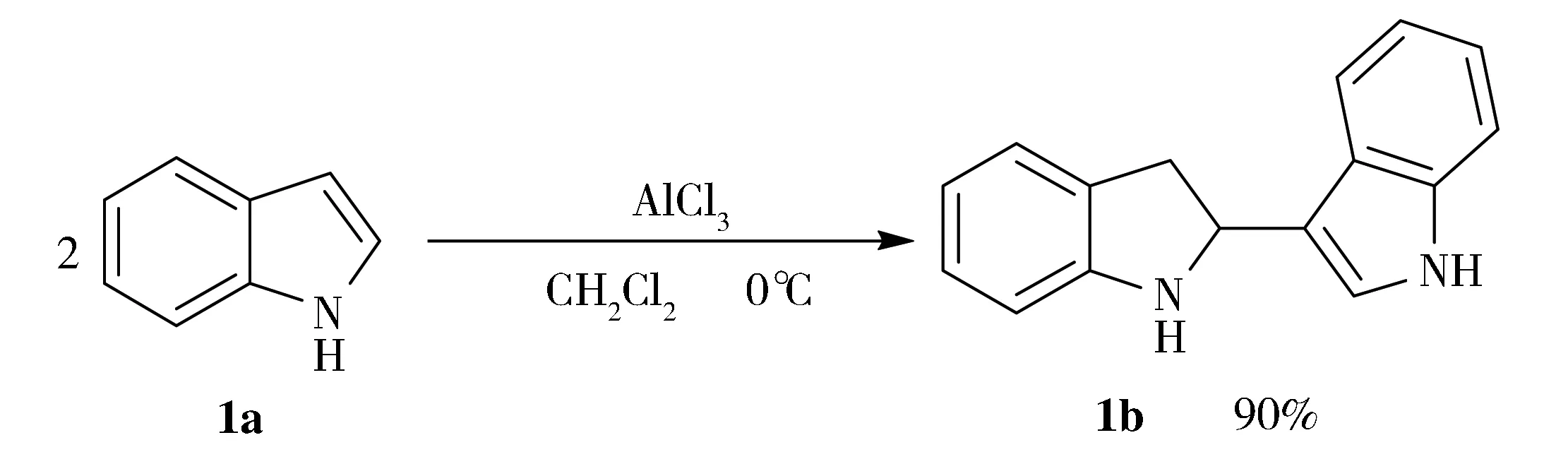


1 实验部分
1.1 主要仪器与试剂
仪器:50 mL圆底烧瓶,磁子,暗箱式紫外分析仪(ZF-20D),INOVA-400MHz核磁共振仪(美国Varian公司),BL 120P型电子分析天平,IKA (RH basic 1), SHZ-D(Ⅲ)型循环水式真空泵,RE-2000A型旋转蒸发仪。
试剂:实验中使用的试剂均为市售分析纯。
1.2 合成步骤
在已放入磁子的50 mL圆底烧瓶中,将10 mmol反应底物溶解于20 mL二氯甲烷中,之后把圆底烧瓶放入冰浴中冷却至0℃;在通风良好且磁子搅拌正常的条件下向圆底烧瓶中分批加入12 mmol[13]无水三氯化铝,确保反应继续在0℃下进行;用TLC薄层色谱分析监测反应,当底物完全反应时(小于40 min),向体系中缓慢注入0℃的水并不断搅拌,直至有机层和水层均变为澄清透亮;用分液漏斗将有机层分离并用饱和食盐水洗涤三次,有机层用无水硫酸钠干燥20 min后过滤,滤液经旋蒸浓缩得到粗产品;粗产品经柱层析(乙酸乙酯/石油醚=1/6) 后得到相应的白色固体产物。
1.3 产物结构表征
氘代试剂为DMSO-d6,核磁共振氢谱选取δ=2.50 ppm作为内标;核磁共振碳谱选取δ=39.5 ppm作为内标。
产物1b(3-(indolin-2-yl)-1H-indole)1H NMR (400 MHz, DMSO-d6)δppm:10.93 (s, 1H), 7.54 (d, J=8.0 Hz, 1H), 7.41 (d, J=8.0 Hz, 1H), 7.29 (s, 1H), 7.13-7.06 (m, 2H), 6.99-6.96 (m, 2H), 6.61-6.58 (m, 2H), 5.98 (s, 1H), 5.17 (t, J=8.0 Hz, 1H), 3.44-3.38 (dd, J=8.0 Hz, 1H), 3.07-3.01(dd, J=8.0 Hz, 1H);13C NMR (100MHz, DMSO-d6)δppm:152.06, 136.87, 128.27, 127.15, 125.71, 124.22, 122.10, 121.13, 119.32, 118.36, 118.24, 116.90, 111.59, 108.12, 55.85, 37.42.
产物2b(5-methyl-3-(5-methylindolin-2-yl)-1H-indole)1H NMR (400 MHz, DMSO-d6) δppm:10.64 (s, 1H), 7.24 (s, 1H), 7.16 (d, J=8.0 Hz, 1H), 7.10 (s, 1H), 6.81 (d, J=8.0 Hz, 1H), 6.76 (s, 1H), 6.67 (d, J=8.0 Hz, 1H), 6.38 (d, J=8.0 Hz, 1H), 5.58 (s, 1H), 4.98 (t, J=8.0 Hz, 1H), 3.24-3.18 (dd, J=8.0 Hz, 1H), 2.90-2.84 (dd, J=8.0 Hz, 1H), 2.25 (s, 3H), 2.09 (s, 3H);13C NMR (100MHz, DMSO-d6) δppm:149.86, 135.21, 128.68, 127.26, 126.61, 126.02, 125.30, 124.97, 122.65, 122.09, 118.91, 117.61, 111.25, 108.08, 56.26, 37.44, 21.36, 20.59.
产物3b(5-bromo-3-(5-bromoindolin-2-yl)-1H-indole)1H NMR (400 MHz, DMSO-d6) δppm: 11.15 (s, 1H), 7.68 (s, 1H), 7.36-7.34 (m, 2H), 7.21-7.17 (m, 2H), 7.09 (d, J=8.0 Hz, 1H), 6.49 (d, J=8.0 Hz, 1H), 6.20 (s, 1H), 5.14 (t, J=8.0 Hz, 1H), 3.41-3.36 (dd, J=8.0 Hz, 1H), 3.01-2.95 (dd, J=8.0 Hz, 1H);13C NMR (100MHz, DMSO-d6) δppm: 151.28, 135.48, 131.11, 129.60, 127.38, 126.92, 123.86, 123.63, 121.52, 117.59, 113.62, 111.12, 109.52, 107.27, 55.77, 37.04 .
2 分析与讨论
2.1 核磁数据分析
首先由核磁数据可知三个产物的碳、氢原子个数:产物1b含有14个氢原子和16个碳原子;产物2b含有18个氢原子和18个碳原子;产物3b含有12个氢原子和16个碳原子。
其次可知三个产物的1H NMR信号是相似的,均有五种相似的特征峰。以产物1b为例说明:

值得注意的是产物2b与1b、3b相比,1H NMR和13C NMR在高场有两个明显的-Me信号。另外,产物2b和3b由于-Me和-Br的特殊位置,使其1H NMR在低场多了两组单峰。
综上所述,三个产物的1H NMR和13C NMR均与其结构相符。
2.2 反应机理讨论
笔者结合相关文献及实验结果,提出了可能的反应机理:首先三氯化铝活化吲哚的C-3位[13],生成亚胺离子中间体[14-15],使吲哚的C-2位显正电性;接下来另一分子吲哚C-3位的碳原子[16]进攻这个正电中心[17],实现偶联;偶联中间体发生质子转移[16]并失去三氯化铝形成最终产物,离去的三氯化铝继续催化反应(图1)。

Fig.1 Possible reaction mechanism图1 可能的反应机理
3 结论
综上所述,本文采用路易斯酸无水三氯化铝代替质子酸或酸酐作催化剂实现了系列吲哚的自身偶联反应。这种新方法与前人工作相比的优势有:(1)催化剂廉价易得且使用方便;(2)反应时间短;(3)产物收率高。文中提出的反应机理为今后吲哚相关偶联反应的研究提供了依据。目前,有关此反应的应用仍在研究当中。
[1] Bandini M,Eichholzer A.Catalytic Functionalization of Indoles in a New Dimension[J].AngewChemIntEd,2009,48:9608-9644.DOI:10.1002/anie.200901843.
[2] Goswami P,Borah A J,Phukan P.Formation of Cyclohepta[b]indole Scaffolds via Heck Cyclization:A Strategy for Structural Analogues of Ervatamine Group of Indole Alkaloid[J].JOrgChem,2015,80:438-446.DOI:10.1021/jo502443a.
[3] Harrison J G,Gutierrez O,Jana N,etal.Mechanism of Rh2(Ⅱ)-Catalyzed Indole Formation:The Catalyst Does Not Control Product Selectivity[J].JAmChemSoc,2016,138:487-490.DOI:10.1021/jacs.5b11427.
[4] Mei Liangyong,Wei Yin,Tang Xiangying,etal.Catalyst-Dependent Stereodivergent and Regioselective Synthesis of Indole-Fused Heterocycles through Formal Cycloadditions of Indolyl-Allenes[J].JAmChemSoc,2015,137:8131-8137.DOI:10.1021/jacs.5b02080.
[5] Ohtaka A,Kozono M,Takahashi K,etal.Linear Polystyrene-stabilized Pt Nanoparticles Catalyzed Indole Synthesis in Water via Aerobic Alcohol Oxidation[J].ChemLett,2016,45:758-760.DOI:10.1246/cl.160331.
[6] Shi Zhuangzhi,Glorius F.Efficient and Versatile Synthesis of Indoles from Enamines and Imines by Cross-Dehydrogenative Coupling[J].AngewChemIntEd,2012,51:9220-9222.DOI:10.1002/anie.201205079.
[7] 李倩倩,李振,秦金贵.新型含吲哚基团的光电功能材料[J].化学进展,2009,21 (12):2578-2588.DOI:1005-281X (2009) 12-2578-11.
[8] Lancianesi S,Palmieri A,Petrini M.Synthetic Approaches to 3-(2-Nitroalkyl) Indoles and Their Use to Access Tryptamines and Related Bioactive Compounds[J].ChemRev,2014,114:7108-7149.DOI:10.1021/cr400676v.
[9] Zhang Zhiwei,Xue Hong,Li Hailing,etal.Collective Synthesis of 3-Acylindoles,Indole-3-carboxylic Esters,Indole-3-sulfinic Acids,and 3-(Methylsulfonyl)indoles from Free (N-H) Indoles via Common N-Indolyl Triethylborate[J].OrgLett,2016,18:3918-3921.DOI:10.1021/acs.orglett.6b01970.
[10] Xu Xiuhua,Liu Guokai,Azuma A,etal.Synthesis of Indole and Biindolyl Triflones:Trifluoromethanesulfonylation of Indoles with Tf2O/TTBP (2,4,6-tri-tert-butylpyridine) System[J].OrgLett,2011,13:4854-4857.DOI:10.1021/ol201931x.
[11] 席真,陈红军,陈文彬.一种吲哚咔唑类化合物及其制备方法和应用:中国,201210104960.3[P].2013-10-23.
[12] 邢其毅,裴伟伟,徐瑞秋,等.基础有机化学[M].(第三版).北京:高等教育出版社,2005:478-479.
[13] Ottoni O,Neder A V F,Dias A K B,etal.Acylation of Indole under Friedel-Crafts Conditions——An Improved Method To Obtain 3-Acylindoles Regioselectively[J].OrgLett,2001,3:1005-1007.DOI:10.1021/ol007056i.
[14] Yamamoto K,Kimura S,Murahashi T.σ-π Continuum in Indole-Palladium(Ⅱ) Complexes[J].AngewChemIntEd,2016,55:5322-5326.DOI:10.1002/anie.201601992.
[15] DiPoto M C,Hughes R P,Wu J.Dearomative Indole (3+2) Reactions with Azaoxyallyl Cations——New Method for the Synthesis of Pyrroloindolines[J].JAmChemSoc,2015,137:14861-14864.DOI:10.1021/jacs.5b10221.
[16] Shiri M.Indoles in Multicomponent Processes (MCPs)[J].ChemRev,2012,112:3508-3549.DOI:10.1021/cr2003954.
[17] Chen Shanping,Li Yuxia,Ni Penghui,etal.Indole-to-Carbazole Strategy for the Synthesis of Substituted Carbazoles under Metal-Free Conditions[J].OrgLett,2016,18:5384-5387.DOI:10.1021/acs.orglett.6b02762.
Study on Homocoupling Reaction of Indoles using Anhydrous Aluminum Chloride as a Catalyst
YU Jingwen,SONG Luna
(Department of Basic Medical Sciences,Jinci College of Shanxi Medical University,Taiyuan 030025,China)
Instead of using proton acid or anhydride as the catalyst,a novel AlCl3-catalyzed homocoupling reaction of indoles was reported herein. This methodology features using low-cost catalyst, simple operation, high yields (86%-90%), and short reaction time (<40 min), which has applications in organic synthesis due to the atomic economic concerns (the atom utilization is 100%). The structure of products were characterized by1H NMR spectra and13C NMR spectra. In addition, a plausible mechanism of this reaction was proposed.
synthetic method;indole;anhydrous aluminum chloride;homocoupling
10.13451/j.cnki.shanxi.univ(nat.sci.).2017.02.011
2016-10-05;
2016-11-10
山西医科大学校级科研基金项目(02201540)
于静文(1988-),男,山西长治人,硕士研究生,实验教师,主要从事新药设计与合成方面的研究工作。E-mail:469165514@qq.com
O626
A
0253-2395(2017)02-0331-05
