RP-HPLC法测定卡博替尼原料药中卡博替尼的含量
2017-05-16詹长娟南京理工大学泰州科技学院环境与制药工程学院江苏泰州225300
徐 伟,詹长娟,王 华,王 翼,郭 琪(南京理工大学泰州科技学院环境与制药工程学院,江苏泰州225300)
RP-HPLC法测定卡博替尼原料药中卡博替尼的含量
徐 伟*,詹长娟#,王 华,王 翼,郭 琪(南京理工大学泰州科技学院环境与制药工程学院,江苏泰州225300)
目的:建立测定卡博替尼原料药中卡博替尼含量的方法。方法:采用反相高效液相色谱法。色谱柱为Inertsil ODS-SP C18,流动相为乙腈-0.02 mol/L乙酸铵缓冲溶液(pH 5.2)(52∶48,V/V),流速为1.0 mL/min,检测波长为241 nm,柱温为38 ,进样量为20 μL。结果:卡博替尼检测质量浓度线性范围为9.88~49.40 μg/mL(r=0.999 9);定量限为11.46 ng,检测限为3.36 ng;精密度、稳定性、重复性试验的RSD<2.0%;加样回收率为98.5~101.7%(RSD=1.2%,n=9)。结论:该方法操作简便、结果准确,可用于卡博替尼原料药中卡博替尼的含量测定。
卡博替尼;反相高效液相色谱法;含量测定
卡博替尼(Cabozantinib)是一种多靶点分子靶向药物,具有显著的抗癌活性,可用于治疗甲状腺髓样癌(Medullary thyroid carcinoma,MTC),开创了MTC靶向治疗的新时代[1-2]。该药于2012年11月由FDA批准上市,用于不可手术切除的恶性局部晚期或转移性MTC的治疗[3-4]。临床试验已证实,其在治疗MTC时具有快速、明确且显著的临床疗效和安全性,其为进展性、转移性MTC患者带来了希望,具有良好的应用前景[5-7]。但目前尚未见测定卡博替尼原料药中卡博替尼含量的报道。因此,笔者采用反相高效液相色谱法(RP-HPLC)建立了测定卡博替尼原料药中卡博替尼的含量,以期为卡博替尼原料药的质量控制提供参考。
1 材料
1.1 仪器
LC-20AT型HPLC仪,包括UV-2450紫外分光光度计、SPD-20A检测器、CTO-10AS VP柱温箱、LC-20AT AB泵、DGU-20A3在线脱气机(日本Shimadzu公司);MJF-300型超声波清洗机(无锡市美极超声波清洗机设备公司,功率:300 W,频率:40 kHz);MP511型实验室pH计(上海精密仪器仪表有限公司)。
1.2 药品与试剂
卡博替尼原料药(南京理工大学泰州科技学院环境与制药工程学院实验室自制,批号:20150101、20150102、20150103);卡博替尼对照品(南京理工大学泰州科技学院环境与制药工程学院实验室自制,批号:20141220,纯度:98.8%);乙腈为色谱纯,其余试剂均为分析纯,水为娃哈哈纯净水。
2 方法与结果
2.1 色谱条件
色谱柱:Inertsil ODS-SP C18(250 mm×4.6 mm,5 μm);流动相:乙腈-0.02 mol/L乙酸铵缓冲溶液(pH 5.2)(52∶48,V/V);流速:1.0 mL/min;检测波长:241 nm;柱温:38 ;进样量:20 μL。
2.2 溶液的制备
2.2.1 对照品溶液 取卡博替尼对照品约50 mg,精密称定,置于500 mL量瓶中,加乙腈定容,称定质量,超声处理10 min使其溶解,放冷,再次称定质量,用乙腈补足减失的质量,摇匀,即得对照品贮备液。取上述对照品贮备液1 mL,置于10 mL量瓶中,加流动相定容,摇匀,即得。
2.2.2 供试品溶液 取样品约50 mg,精密称定,置于500 mL量瓶中,加乙腈定容,称定质量,超声处理10 min使其溶解,放冷,再次称定质量,用乙腈补足减失的质量,摇匀,即得供试品贮备液。取上述供试品贮备液1 mL,置于10 mL量瓶中,加乙腈定容,摇匀,即得。
2.2.3 空白溶液 取流动相适量作为空白溶液。
2.3 系统适用性试验
精密量取“2.2”项下对照品溶液、供试品溶液和空白溶液各适量,按“2.1”项下色谱条件进样测定,记录色谱,详见图1。由图1可知,在该色谱条件下,各成分均能达到基线分离,分离度>1.5;理论板数以卡博替尼峰计为13 698,保留时间为16.6 min。结果表明,其他成分对测定无干扰。
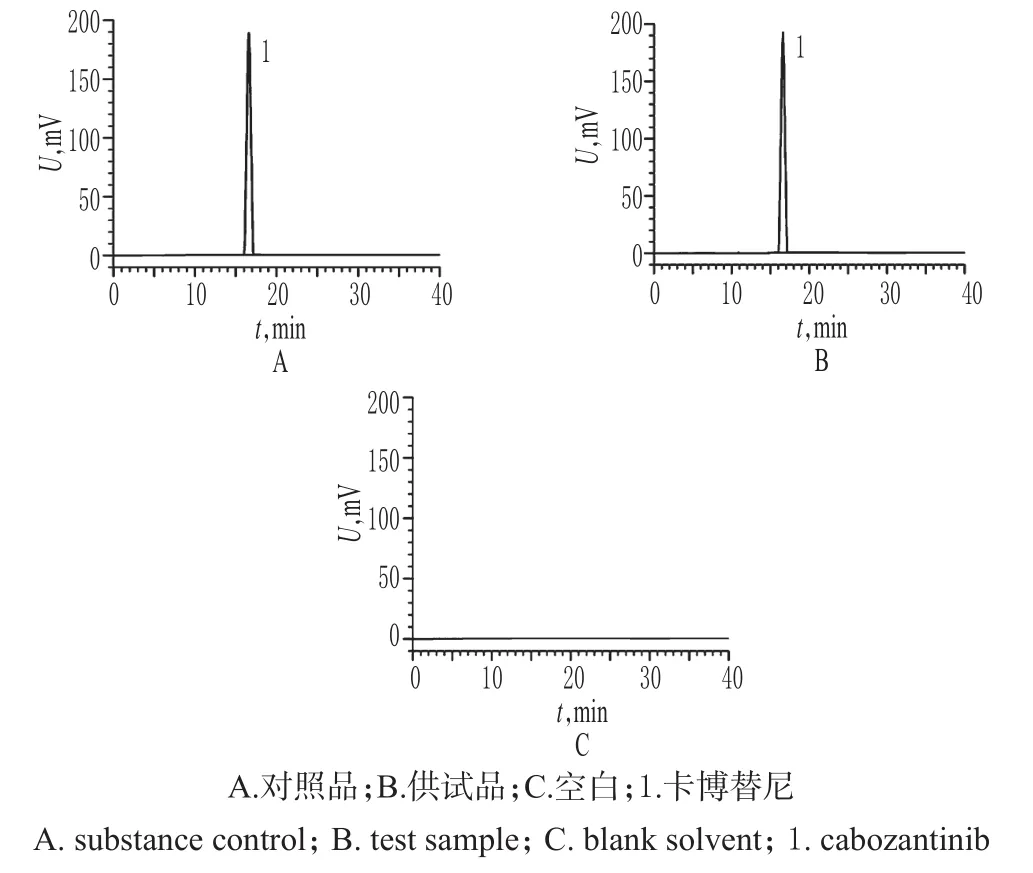
图1 高效液相色谱图Fig 1 HPLC chromatograms
2.4 线性关系考察
精密量取“2.2.1”项下对照品贮备液1.0、2.0、3.0、4.0、5.0 mL,分别置于10 mL量瓶中,加乙腈定容,摇匀,即得系列对照品溶液。取上述系列对照品溶液各20 μL,按“2.1”项下色谱条件进样测定,记录峰面积。以卡博替尼质量浓度(x,μg/mL)为横坐标、峰面积(y)为纵坐标进行线性回归,得卡博替尼回归方程为y=1.319× 108x+4.126×103(r=0.999 9)。结果表明,卡博替尼检测质量浓度线性范围为9.88~49.40 μg/mL。
2.5 定量限与检测限考察
取“2.2.1”项下对照品溶液适量,倍比稀释,按“2.1”项下色谱条件连续进样测定6次,记录峰面积。当信噪比为10∶1时,得定量限(LOQ);当信噪比为3∶1时,得检测限(LOD)。结果,卡博替尼的LOQ为11.46 ng,LOD为3.36 ng。
2.6 精密度试验
取“2.2.1”项下对照品溶液20 μL,按“2.1”项下色谱条件连续进样测定6次,记录峰面积。结果,卡博替尼峰面积的RSD=1.7%(n=6),表明仪器精密度良好。
2.7 稳定性试验
取“2.2.2”项下供试品溶液(批号:20150101)适量,分别于室温下放置0、2、4、6、8 h时按“2.1”项下色谱条件进样测定,记录峰面积。结果,卡博替尼峰面积的RSD=1.4%(n=5),表明供试品溶液在室温放置8 h内稳定性良好。
2.8 重复性试验
取样品(批号:20150101)适量,共6份,按“2.2.2”项下方法制备供试品溶液,再按“2.1”项下色谱条件进样测定,记录峰面积并计算样品含量。结果,卡博替尼的平均含量为97.8%,RSD=1.4%(n=6),表明本方法重复性良好。
2.9 加样回收率试验
取样品(批号:20150101)约10 mg,共9份,分别置于100 mL量瓶中,加入低、中、高质量的卡博替尼对照品,按“2.2.2”项下方法制备供试品溶液,再按“2.1”项下色谱条件进样测定,记录峰面积并计算加样回收率,结果见表1。
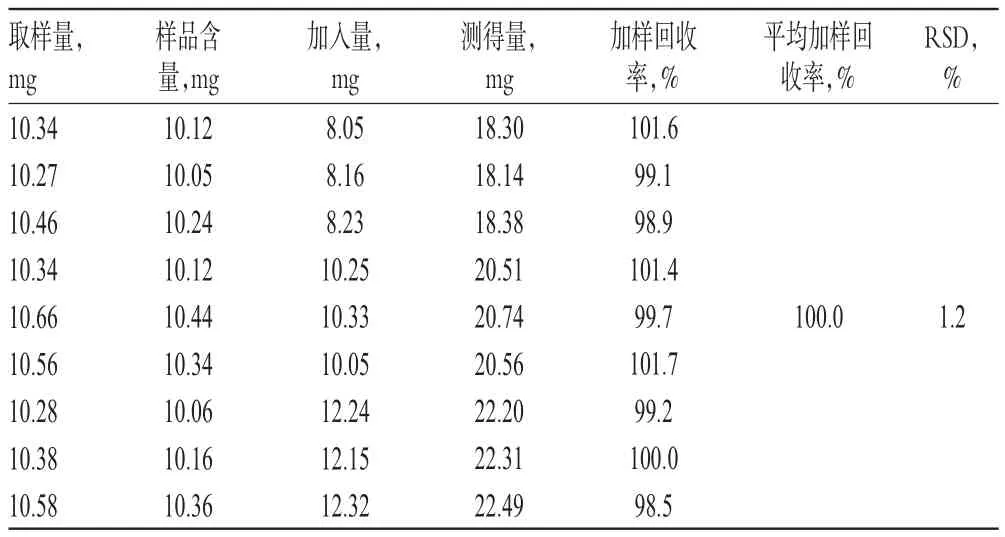
表1 加样回收率试验结果(n=9)Tab 1 Result of recovery test(n=9)
2.10 样品含量测定
取3批样品(批号:20150101、20150102、20150103)各适量,分别按“2.2.2”项下方法制备供试品溶液,再按“2.1”项下色谱条件进样测定,记录峰面积并计算卡博替尼的含量。结果,3批样品中卡博替尼的含量分别为97.9%、98.3%、98.6%(n=3)。
3 讨论
笔者曾考察乙腈-水、乙腈-0.02 mol/L乙酸铵缓冲溶液(pH5.2)作为本研究的流动相时对色谱峰分离的影响。结果,当乙腈-水为流动相时,调节流动相的比例,各色谱峰分离均不理想;当乙腈-0.02 mol/L乙酸铵缓冲溶液(pH5.2)为流动相时,乙腈与0.02 mol/L乙酸铵缓冲溶液体积比在45∶55~45∶55之间时各色谱峰均可达到基线分离,且乙腈比例越高,保留时间越小,因此选择乙腈-0.02 mol/L乙酸铵缓冲溶液(pH5.2)(52∶48,V/V)为本试验的流动相。
综上所述,本方法操作简便、结果准确,可用于卡博替尼原料药中卡博替尼的含量测定。
[1] Bowles DW,Kessler ER,Jimeno A.Multitargeted tyrosine kinase inhibitors in clinical development:focus on XL-184(cabozantinib)[J].Drugs Today:Barc,2011,47 (11):857-868.
[2] 张秀颖,刘尧,白秋江,等.新型分子靶向抗癌药物卡博替尼[J].医药导报,2013,32(11):1468-1470.
[3] 夏训明.美国FDA批准Cometriq(cabozantinib)治疗甲状腺髓样癌[J].广东药学院学报,2012,28(6):627.
[4] 陈惠玲,张志叶,杨彦彪.治疗转移性甲状腺髓样癌的新药:Cabozantinib[J].中国药房,2014,25(29):2766-2769.
[5] 郑希元,姜汉杰,蒲小平.抗甲状腺髓样癌新药卡博替尼[J].中国新药杂志,2013,22(17):1990-1993.
[6] 吕娟丽,刘洋,孙慧萍.甲状腺髓样癌治疗新药Cabozantinib苹果酸盐[J].中国临床药学杂志,2014,23(1):28-62.
[7] 赵锐,张喜全,孟庆义.以c-Met为靶点的小分子酪氨酸激酶抑制剂的研究进展[J].药物与临床研究,2012,20 (6):519-522.
(编辑:刘 柳)
Content Determination of Cabozantinib in Its Raw Material by RP-HPLC
XU Wei,ZHAN Changjuan,WANG Hua,WANG Yi,GUO Qi(School of Environmental and Pharmaceutical Engineering,Taizhou Institute of Sci.&Tech.,Nanjing University of Science&Technology,Jiangsu Taizhou 225300,China)
Cabozantinib;RP-HPLC;Content determination
R917
A
1001-0408(2017)12-1696-03
2016-04-30
2017-01-12)
*讲师,硕士。研究方向:药物化学。电话:0523-86150675。E-mail:541175674@qq.com
#通信作者:讲师,硕士。研究方向:药物分析。E-mail:Jenny_yz@sina.com
DOI10.6039/j.issn.1001-0408.2017.12.31
ABSTRACTOBJECTIVE:To establish a method for the content determination of cabozantinib in its raw material.METHODS:RP-HPLC method was adopted.The determination was performed on Inertsil ODS-SP C18column with mobile phase consisted of acetonitrile-0.02 mol/L ammonium acetate buffer(pH 5.2,52∶48,V/V)at the flow rate of 1.0 mL/min.Detection wavelength was set at 241 nm,the column temperature was 38 ,and sample size was 20 μL.RESULTS:The linear range of cabozantinib were 9.88-49.40 μg/mL(r=0.999 9).The limit of quantitation was 11.46 ng,and the limit of detection was 3.36 ng.The RSDs of precision,stability,repeatability tests were all lower than 2.0%;recoveries were 98.5%-101.7%(RSD=1.2%,n=9).CONCLUSIONS:The method is simple,accurate and suitable for the content determination of cabozantinib in its raw material.
