Ag3XO4(X=P,As,V)电子结构及光催化性质的第一性原理计算
2017-05-11李蛟陈忠
李 蛟 陈 忠
(1山东理工大学材料科学与工程学院,山东淄博255049;2南洋理工大学材料科学与工程学院,新加坡639798)
Ag3XO4(X=P,As,V)电子结构及光催化性质的第一性原理计算
李 蛟1,2,*陈 忠2
(1山东理工大学材料科学与工程学院,山东淄博255049;2南洋理工大学材料科学与工程学院,新加坡639798)
基于密度泛函理论的第一性原理对Ag3XO4(X=P,As,V)电子结构及光催化性质进行了对比研究。与Ag3PO4相比,Ag3VO4较好的光催化稳定性主要源于其结构中Ag―O间较强的作用力增加了对Ag+的控制,而Ag3VO4弱的光催化活性与其导带底中存在d轨道成份以及较低的价带边势(2.335 V,vs NHE)有关;对Ag3AsO4而言,其优于Ag3PO4光催化活性的原因基于三个方面:(1)由高分散Ag s-Ag s杂化轨道构成的导带底能带;(2)窄的带隙(1.91 eV);(3)宽的可见光响应范围以及高的光吸收系数。此外,Ag3XO4(X=P,As, V)均为间接带隙半导体光催化材料,其中,Ag3VO4有用于分解水制氢研究的可能;上述计算结果与实验结果吻合。
第一性原理;Ag3XO4(X=P,As,V);能带结构;态密度;光催化
1 引言
磷酸银(Ag3PO4)是一种具有高催化活性的可见光催化材料。在波长小于530 nm光源照射下,Ag3PO4能够表现出强的光催化氧化性能以及降解有机污染物的能力,量子效率超过90%(AgNO3作牺牲剂)1。对于Ag3PO4高催化活性的原因,朱永法等2认为,除了Ag3PO4高的价带位置使价带上的空穴具有很强的氧化能力外,Ag3PO4导带中离域的π*键以及的诱导效应也起到了重要作用。针对这一问题,Umezawa等3则提出不同观点。他们采用密度泛函理论计算分析后认为,Ag3PO4内部PO4四面体单元中牢固P―O键会减弱Ag―O键的共价特性,从而抑制了Ag d轨道与O p轨道的杂化,减少了导带底(CBM)中d轨道的成分,导致高分散性Ag s-Ag s杂化轨道的形成,而CBM中高度分散的能带结构有利于电子-空穴分离,因而Ag3PO4能够表现出优异的光催化活性。尽管上述二种观点表述重点有所不同,但均未否认PO()结
4构单元在Ag3PO4高光催化活性中所起到的重要作用。基于上述认知,部分研究人员对含有XO4结构单元的其它银基半导体光催化材料产生了浓厚兴趣,并陆续开展了一系列卓有成效的研究工作4-8。
硫属化合物具有好的可见光响应特性及适合的带边位置,常用于裂水制氢研究9,10。以某硫属元素,如S、Se完全取代Ag3PO4中的O而得到的材料是否具有高的光催化活性?结合这一问题,Ma等4利用第一性原理系统研究了Ag3PSe4与Ag3PS4的电子结构与光催化性质。构建模型显示Ag3PSe4与Ag3PS4晶胞结构中存在XO4(X=S,Se)正四面体单元。结果表明,Ag3PSe4具有比Ag3PO4更窄的带隙(2.09 eV)以及对可见光高的吸收系数,同时,CBM明显低于H+/H2的还原电势表明了Ag3PSe4的裂水制氢性能;而Ag3PS4带隙为2.88 eV,说明其亦具有可见光响应特性,而更低的CBM位置(-0.53 V,vs NHE)意味着Ag3PS4的裂水制氢能力会更高。然而,由于实验制备条件要求较为苛刻11,12,目前关于Ag3PSe4与Ag3PS4光催化性能的实验研究工作还鲜见报道。与S/Se对O的取代不同,As/V取代P―O中P而得到化合物Ag3AsO4/Ag3VO4的实验研究工作已多有开展。Tang等5首次通过沉淀法制备出Ag3AsO4,发现Ag3AsO4在可见光照射下光催化降解罗丹明B与甲基橙的能力要优于Ag3PO4。Reunchan等13将上述现象的产生归因于Ag3AsO4具有小的电子有效质量与低的价带边位置。对于Ag3VO4而言,一般实验条件下获得的Ag3VO4光催化活性不高,这与其内部极低的空穴浓度有很大关系14,但可通过减小颗粒尺寸、增大比表面积、掺杂或复合等实验手段予以改善6-8。不过与Ag3PO4相比,Ag3VO4较好的光催化稳定性说明该材料在光催化领域依然具有一定的研究参考价值15。对比Ag3VO4与Ag3AsO4光催化性能的实验研究工作,涉及Ag3VO4与Ag3AsO4光催化性能的理论研究工作才刚刚起步13,16。对比Ag3PO4,Ag3VO4与Ag3AsO4表现出的某些特殊光催化性能,如Ag3AsO4优异的光催化活性、Ag3VO4良好的光催化稳定性依然需要进行系统而深入的探索性研究。
鉴于上述问题,本文拟利用基于密度泛函理论(DFT)的第一性原理,采用规范-守恒赝势平面波方法,通过对Ag3XO4(X=P,As,V)系列银基材料键布居、能带结构、态密度以及带边势与光学性质的计算,以Ag3PO4参比,对比分析Ag3AsO4与Ag3VO4电子结构及光催化性质,从理论上解释Ag3AsO4/Ag3VO4所表现出的特殊光催化性能。上述研究结果的获得对于高活性、高稳定性银基光催化材料结构设计具有较强的指导意义。
2 模型构建与计算方法
2.1 模型构建
本文构建Ag3XO4(X=P,As,V)晶胞模型如图1所示。其中,Ag3XO4(X=P,As)为体心立方晶型,属P43n空间群(218),模型构建所用晶格常数与原子分数坐标分别为:Ag3PO4,a=b=c=0.6004 nm,α=β=γ=90°,Ag(0.25 0 0.5),P(0 0 0),O (0.148 0.148 0.148)17;Ag3AsO4,a=b=c=0.6120 nm,α=β=γ=90°;Ag(0.25 0 0.5),As(0 0 0),O(0.335 0.335 0.335)18。Ag3VO4为单斜晶型,属I42m空间群(122),其模型构建所用晶格常数与原子分数坐标为:a=b=0.4997 nm,c=0.9691 nm,α=β=γ=90°;Ag1(0 0.5 0.75),Ag2(0 0 0.5),V(0 0 0),O(0.8073 0.1927-0.0997)19。从图1可以看出,Ag3XO4(X=P,As,V)晶胞结构中均存在XO4(X=P,As,V)正四面体单元。
2.2 计算方法
本文对Ag3XO4(X=P,As,V)电子和光学性质的计算采用基于密度泛函理论的规范-守恒赝势平面波方法,平面波截断能Ecut=750 eV,电子交换相关项采用杂化泛函PBE0。迭代过程中能量自洽收敛精度设置为1.0×10-5eV·atom-1,作用在每个原子上的力不大于0.3 eV·nm-1,内应力不大于0.05 GPa,原子之间的距离不大于1.0×10-4nm,计算第一布里渊区时的k点分割设置针对不同研究对象分别为4×4×4、4×4×4以及3×3×3。计算所考察各研究化合物价电子设置分别为:Ag3PO4,Ag-4d105s1,P-3s23p3,O-2s22p4;Ag3AsO4,Ag-4d105s1, As-4s24p3,O-2s22p4;Ag3VO4,Ag-4d105s1,V-3s23p3, O-2s22p4,其余轨道电子作为苾电子进行计算。在计算电子和光学性质之前,用GGA/PBE对构建晶胞模型进行几何优化,以获得最优结构。
3 结果与讨论
3.1 结构优化分析
Ag3XO4(X=P,As,V)晶格常数优化结果如表1所示。对比发现,各模型优化后晶格常数与相应的实验数值非常接近,其相关误差在0.14%(a, Ag3VO4)-1.45%(a,Ag3AsO4)之间,小于允许误差2%,表明本文计算方法设计合理,结果可靠。
3.2 键布居分析
键布居是一种可以用来分析化学键特性的常用参数,键布居数值越大,化学键共价特性越强,反之则表现出更强的离子键特征20,21。表2为本文计算得到的不同材料中X―O(X=P,As,V)与Ag―O键布居及键长。O―P、O―As与O―V键布居值分别为0.62、0.56与0.67,说明与O―P相比,O―As离子键特性增强,而O―V共价键特性增强。对比键长发现,O―As与O―V键长均大于O―P,这应该与As5+/V5+较大的离子半径有关。另一方面,Ag3XO4(X=P,As,V)中Ag―O键长分别为0.2407,0.2392 nm以及0.2337/0.2294 nm。键长变短说明P被As或V取代后,Ag3XO4(X=As,V)中Ag―O间作用力逐渐增强。基于文献理论3,Ag3PO4中Ag―O间作用力受PO4四面体单元中P―O间强作用力的负面影响,Ag―O间作用力增强只能说明Ag3XO4(X=As,V)中O―X(X=As,V)间作用力弱于O―P键。然而,与Ag3PO4相比,Ag3AsO4中Ag―O键长仅减小了0.0015 nm,说明O―As与O―P间作用力差别不大。多个研究小组证实,Ag3PO4光稳定性差主要源于Ag+的不稳定22,23,而Ag―O间作用力增强势必会提高对Ag+的控制力,提升Ag+的稳定性。Ag3XO4(X=P,As,V)系列银基材料中Ag3VO4的Ag―O键作用力最强,表明Ag3VO4理论上应具有良好的光催化稳定性,这与吴尧15的实验研究结论吻合。
3.3 电子结构分析
图2-4为Ag3XO4(X=P,As,V)的能带结构图,费米能级始终定为0点(EF=0 eV)。对比图2 (a)、图3(a)可知,Ag3PO4与Ag3AsO4能带结构非常相似,其价带顶与导带底均分别位于布里渊区M点与G点,这表明Ag3P(As)O4均属于间接带隙半导体材料。此外,进一步对比观察Ag3PO4与Ag3AsO4费米能级附近能带图(图2(b)与图3(b))发现,二者位于价带顶及导带底的能带具有相似的变化态势:位于G点的导带底能带均具有明显抛物线形态,曲率较大,表现出各向同性特征,而位于M点的价带顶能带则都呈现出相对平缓形态,其变化显示各向异性特征。通常情况下,依据能带结构中价带顶或导带底能带变化趋势即可初步判断出价带顶空穴或导带底电子的有效质量,并对相关载流子的迁移能力进行衡量24,25。综合上述分析说明Ag3P(As)O4中位于导带底的光生电子有效质量均要轻于价带顶上的空穴有效质量,且均具有各向同性特征,非局域性程度大,载流子易在材料内部迁移,如果再考虑到间接带隙半导体中光生电子与空穴无法直接复合26,27,因此,Ag3P(As)O4均应具有光生电子-空穴复合几率低,电子迁移速率高等特点。
Ag3PO4与Ag3AsO4不同之处在于其带隙大小。Ag3PO4(间接)带隙2.49 eV,这一计算结果高于文献1的实验测试结果(2.36 eV),但与Ma等6得到的计算结果(2.49 eV)完全相同。这种理论计算与实验之间带隙偏差的形成与泛函本身的计算特征有关,但不会影响对研究对象电子结构及相关性质的理论分析28,29。与Ag3PO4相比,Ag3AsO4带隙仅为1.91 eV,高于文献7的测试结果(1.60 eV)。较低的带隙意味着光生电子从价带至导带具有更高的跃迁概率,这应该是Ag3AsO4具有相对较高光催化活性的原因之一。
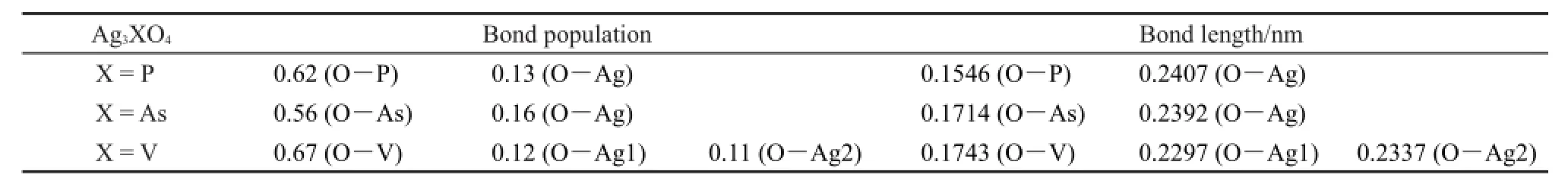
表2 Ag3XO4(X=P,As,V)的键布居和键长Table 2 Calculated bond population and bond length of Ag3XO4(X=P,As,V)
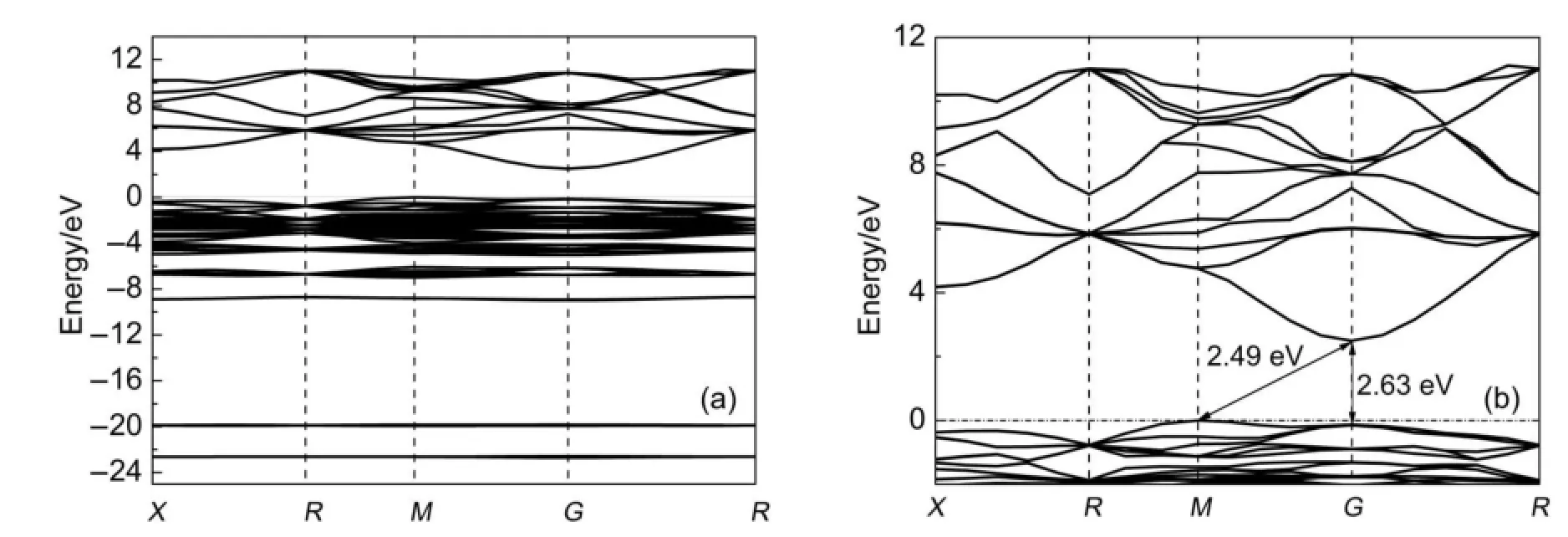
图2 Ag3PO4的能带结构(a)以及费米能级附近能带结构(b)Fig.2(a)Band structure of Ag3PO4and(b)magnified band structure around the Fermilevel
图4 显示了Ag3VO4的能带结构。由于价带顶位于布里渊区P点,而导带底位于布里渊区G点,因此Ag3VO4也是一种间接带隙半导体材料。Ag3VO4带隙计算结果为2.39 eV,低于文献14的计算结果(2.60 eV),但略高于文献30的实验测试结果(2.26 eV)。考虑到Ag3VO4带隙高于Ag3AsO4,低于Ag3PO4,说明Ag3VO4接受光子能量产生电子与空穴的能力应介于Ag3AsO4与Ag3PO4之间,然而,单一Ag3VO4并未表现出相应高的光催化活性31,显然肯定有其它因素负面影响了Ag3VO4的光催化性能。
通过态密度分析可以获取化合物中各原子电子态对导带与价带的贡献信息32。图5给出了Ag3XO4(X=P,As,V)总态密度(TDOS)与不同原子亚层轨道分态密度(PDOS)分布情况。Ag3PO4与Ag3AsO4态密度图相似度很高(图5(a,b))。在价带低能区间(E<-15 eV),Ag3P(As)O4态密度主要由O 2s、P 3s(As 4s)以及P 3p(As 4p)态构成,较低能区间(-15 eV≤E<-5 eV)主要由O 2p、P 3s(As 4s)以及P 3p(As 4p)态构成,而在近费米能级价带区(-5 eV≤E<0),两种材料态密度分布则完全一致,主要由O 2p与Ag 4d态构成,P 3p或As 4p贡献很少;另一方面,导带靠近费米能级区(0<E≤7 eV)的态密度主要由Ag 5s态贡献,P(As)与O的s、p轨道电子贡献几乎不存在。而在导带高能区(7 eV<E≤15 eV),Ag3PO4与Ag3AsO4的态密度还是有不同之处,Ag3PO4主要由Ag 5s、Ag 5p以及少量P 3p构成,而对于Ag3AsO4,除Ag 5s、Ag 5p外,还有较为明显的As 4s,As 4p态。
综合上述分析结果可知,Ag3P(As)O4费米能级附近总态密度几乎完全来自于O 2p,Ag 4d以及Ag 5s态密度的贡献,说明Ag3P(As)O4中载流子输送性质主要由O 2p,Ag 4d以及Ag 5s态电子决定,与P或As原子无关。此外,同Ag3PO4一样,Ag3AsO4导带低是由高分散的Ag s-Ag s杂化轨道组成,这应该是其具有高催化活性的原因之一。另一方面,Ag3P(As)O4价带顶位置的Ag 4d与O 2p态密度均存在部分重叠现象,说明Ag 4d与O 2p轨道之间存在一定杂化作用,但在价带低能区,O 2s与P 3p (As 4p)态密度高度重合,说明O 2s与P 3p或As 4p轨道杂化作用非常强烈,证明在Ag3P(As)O4中O―P(As)键合力要强于Ag―O键。

图3 Ag3AsO4的能带结构(a)以及费米能级附近能带结构(b)Fig.3(a)Band structure of Ag3AsO4and(b)magnified band structure around the Fermilevel
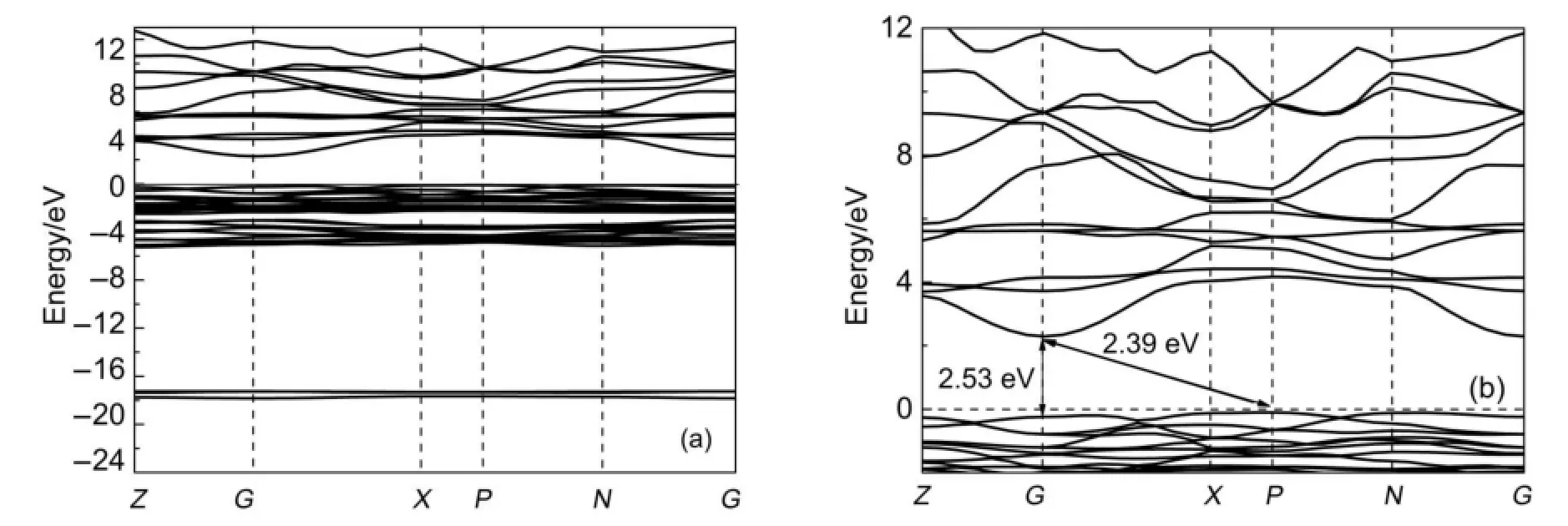
图4 Ag3VO4的能带结构(a)以及费米能级附近能带结构(b)Fig.4(a)Band structure of Ag3VO4and(b)magnified band structure around the Fermilevel
由于V原子d能级的存在,导致Ag3VO4态密度图明显不同与Ag3PO4。从图5(c)可以看出,Ag3VO4导带靠近费米能级区(0<E≤7 eV)的态密度由Ag 5s、V 3d以及O 2p、Ag 5d多种轨道态杂化构成;而在靠近费米能级价带区(-5 eV≤E<0),Ag 4d、V 3d以及O 2p杂化构成Ag3VO4低能价带。与s轨道相比,d轨道对电子的离域性较弱1,因此,Ag3VO4导带底中d轨道成分的增加,必会造成跃迁至导带底的光生电子在材料内部迁移能力下降,这应该是单一Ag3VO4光催化活性不高的主因。此外,与Ag3P(As)O4相比,Ag3VO4态密度图中,O 2s与V 4p或V 3d态密度重合程度要弱于O 2s与P 3p或As 4p间的重合,说明O 2s与V 4p或V 3d轨道杂化作用相对较弱,O―V间作用力要弱于O―P与O―As,这与键布居分析结论是一致的。

图5 (a)Ag3PO4,(b)Ag3AsO4与(c)Ag3VO4总态密度以及各原子亚层轨道态密度图Fig.5 Calculated totaldensity of states(TDOS)and partialdensity of states(PDOS)of (a)Ag3PO4,(b)Ag3AsO4and(c)Ag3VO4
3.4 带边势分析
理想的光催化材料不仅要有合适的能带结构,同时还要有位置恰当的带边势,因为材料导带底与价带顶对应的氧化还原电势能够直接决定材料光催化效率的高低9,33。通常情况下,材料带边对应的氧化还原电势(相对于标准电极电位的氧化还原势)可通过公式(1)、(2)计算获得34,35:

其中,Ec、Ev分别为异带底与价带顶对应的氧化还原电势(相对于标准电极电位的氧化还原势),χ是构成材料各原子密立根电负性几何平均值,Ee为以氢为标准时自由电子的电势(4.50 V,vs NHE),Eg为计算得到的禁带宽度。
分析计算结果可知,尽管Ag3PO4具有高的光催化活性,但是却无法将水还原成氢,原因在于其导带边势(0.215 V,vs NHE)高于H+/H2的还原电势(图6)。与Ag3PO4相似,Ag3AsO4亦具有高的导带边势(0.465 V,vs NHE),说明其同样不具有裂水制氢的能力。然而,Ag3VO4导带边势(-0.055 V(vs NHE))略低于析氢电位,说明此类材料有用于裂水制氢研究的可能。另一方面,相比于Ag3PO4高的价带边势(2.705 V(vs NHE)),Ag3AsO4与Ag3VO4价带边势分别为2.375 V(vs NHE)、2.335 V(vsNHE),说明Ag3As(V)O4中价带空穴的氧化能力均弱于Ag3PO4。
3.5 吸收光谱分析
通常情况下,线性响应范围内固体宏观光学性质可由复介电函数ε(ω)=ε1(ω)+iε2(ω)予以描述,其中,实部ε1(ω)、虚部ε2(ω)以及吸收系数α(ω)等均可以通过Kramers-Krönig色散关系导出36,37。本文计算得到的Ag3XO4(X=P,As,V)吸收光谱如图7所示。Ag3PO4在500 nm以下波段有响应信号,且对光的吸收强度随波长增加而逐渐减小,最终趋于零,这一变化趋势与文献1测试结果吻合。与Ag3PO4相比,Ag3As(V)O4光谱吸收边均发生红移,其中,Ag3AsO4红移程度明显高于Ag3VO4,表明Ag3AsO4具有相对较宽的可见光响应范围,这意味着此类材料中光激电子完成从价带至导带的跃迁只需较小的能量即可实现。另一方面,Ag3As(V)O4在各自光响应范围内对光的吸收强度明显高于Ag3PO4,其中,Ag3AsO4在280-650 nm范围内对光的吸收强度最大,而Ag3VO4在低于280 nm波段具有最大的光吸收系数。

图6 计算的Ag3XO4(X=P,As,V)导带边势与价带边势Fig.6 Calculated CBMand VBMpotentials of Ag3XO4(X=P,As,V) CBM:conduction band minimum;VBM:valence band maximum

图7 Ag3XO4(X=P,As,V)的吸收系数Fig.7 Calculated absorption coefficient of Ag3XO4(X=P,As,V)
4 结论
运用第一性原理方法对Ag3XO4(X=P,As,V)电子结构与光催化性质进行了理论研究。结果表明,与Ag3PO4相比,Ag3VO4结构中Ag―O间作用力增强,O2-对Ag+的控制力增加,改善了Ag+稳定性,Ag3VO4因而表现出较好的光催化稳定性;而Ag3VO4弱的光催化活性与其导带底中存在d轨道成份以及较低的价带边势(2.335 V,vs NHE)有关;对Ag3AsO4而言,其优于Ag3PO4光催化活性的原因主要基于三个方面:①由高分散Ag s-Ag s杂化轨道构成的导带底结构,②更窄的带隙(1.91 eV),③更宽的可见光响应范围以及高的光吸收系数;Ag3XO4(X=P,As,V)均为间接带隙半导体材料,其中,Ag3VO4有用于裂水制氢研究的可能;上述计算结果与实验结果吻合。
(1)Yi,Z.G.;Ye,J.H.;Kikugawa,N.;Kako,T.;Ouyang,S.X.; Stuart-William,H.;Yang,H.;Cao,J.Y.;Luo,W.J.;Li,Z.S.; Liu,Y.;Ray,L.Nat.Mater.2011,9(42),559.doi:10.1038/ NMAT2780
(2)Ma,X.G.;Lu,B.;Li,D.;Shi,R.;Pan,C.S.;Zhu,Y.F.J.Phys. Chem.C 2011,115(11),4680.doi:10.1021/jp111167u
(3)Umezawa,N.;Ouyang,S.;Ye,J.H.Phys.Rev.B 2011,83(3), 287.doi:10.1103/PhysRevB.83.035202
(4)Ma,Z.J.;Yi,Z.G.;Sun,J.;Wu,K.C.J.Phys.Chem.C 2012, 116(47),25074.doi:10.1021/jp3093447
(5)Tang,J.T.;Liu,Y.G.;Li,H.Z.;Tan,Z.;Li,D.T.Chem. Commun.2013,49(48),5498.doi:10.1039/c3cc41090k
(6)Zhang,L.;He,Y.M.;Ye,P.;Qin,W.H.;Wu,Y.;Wu,T.H. Mater.Sci.Eng.B 2013,178(1),45.doi:10.1016/j. mseb.2012.10.011
(7)Tao,X.C.;Hong,Q.;Xu,T.Z.;Liao,F.J.Mater.Sci.-Mater. Electron.2014,25(8),3480.doi:10.1007/s10854-014-2042-8
(8)Wangkawong,K.;Phanichphant,S.;Tantraviwat,D.; Inceesungvorn,B.J.Colloid Interface Sci.2015,454(2044), 210.doi:10.1016/j.jcis.2015.05.025
(9)Savio,J.A.M.;Stephen,A.S.;David,J.M.;Zheng,X.G.; Tang,J.W.Energ.Environ.Sci.2015,8(3),731.doi:10.1039/ C4EE03271C
(10)Yu,X.L.;Du,R.F.;Li,B.Y.;Zhang,Y.H.;Liu,H.J.;Qu,J. H.;An,X.Q.Appl.Catal.B-Environ.2016,182,504. doi:10.1016/j.apcatb.2015.09.003
(11)Andrae,H.;Blachnik,R.J.Therm.Anal.1989,35(2),595. doi:10.1007/BF01904461
(12)Ma,H.W.;Guo,G.C.;Chen,W.T.;Deng,L.;Zhou,G.W.;Dong,Z.C.;Huang,J.S.Chin.J.Struct.Chem.2003,22(2), 161.doi:10.14102/j.cnki.0254-5861.2003.02.008
(13)Reunchan,P.;Boonchun,A.;Umezawa,N.Phys.Chem.Chem. Phys.2016,18(13),23407.doi:10.1039/c6cp03633c
(14)Trimarchi,G.;Peng,H.;Im,J.;Freeman,A.J.;Cloet,V.;Raw, A.;Poeppelmeier,K.R.;Biswas,K.;Lany,S.;Zunger,A.Phys. Rev.B 2011,84(16),3990.doi:10.1103/PhysRevB.84.165116
(15)Wu,X.Synthesis of Ag3VO4and Its Visible-Light Photocatalytic Performance.M.S.Dissertation,Xiangtan University,Xiangtan, 2012.[吴尧.Ag3VO4的制备及可见光光催化性能研究[D].湘潭:湘潭大学,2012.]
(16)Wang,S.M.;Guan,Y.;Wang,L.P.;Zhao,W.;Yang,S.G.;He, H.;Sun,C.J.Comput.Theor.Nanos.2015,12(12),5016. doi:10.1166/jctn.2015.4087
(17)Ng,H.N.;Calvo,C.;Faggiani,R.Acta Crystallogr.Sect.B 1978,34(33),898.doi:10.1107/S0567740878014570
(18)Helmholtz,L.;Levine,R.J.Am.Chem.Soc.1942,64(2),354. doi:10.1021/ja01254a036
(19)Dinnebier,R.E.;Kowalevsky,A.;Reichertt,H.;Jansen,M.New Cryst.St.2007,222(8),420.doi:10.1524/zkri.2007.222.8.420
(20)Qu,L.F.;Hou,Q.Y.;Zhao,C.W.Acta Phys.Sin.2016,65(3), 037103.[曲灵丰,侯清玉,赵春旺.物理学报,2016,65(3), 037103.]doi:10.7498/aps.65.037103
(21)Mayer,I.;Räther,G.;Suhai,S.Chem.Phys.Lett.1998,293(1-2),81.doi:10.1016/S0009-2614(98)00774-X
(22)Yang,Z.M.;Huang,G.F.;Huang,W.Q.;Wei,J.M.;Yan,X. G.;Liu,Y.Y.;Jiao,C.;Wan,Z.;Pan,A.L.J.Mater.Chem.A 2014,2(6),1750.doi:10.1039/c3ta14286h
(23)Cai,W.W.;Li,J.;Zhang,H.Mater.Sci.Technol.2016,24(5), 47.[蔡维维,李蛟,张华.材料科学与工艺,2016,24(5), 47.]doi:10.11951/j.issn.1005-0299
(24)Wu,R.X.;Liu,D.J.;Yu,Y.;Yang,T.Acta Phys.Sin.2016,65 (2),027101.[吴若熙,刘代俊,于洋,杨涛.物理学报, 2016,65(2),027101.]doi:10.7498/aps.65.027101
(25)Huang,D.;Ju,Z.P.;Li,C.S.;Yao,C.M.;Guo,J.Acta Phys. Sin.2014,63(24),247101.[黄丹,鞠志萍,李长生,姚春梅,郭进.物理学报,2014,63(24),247101.]doi:10.7498/ aps.63.247101
(26)Zhang,J.F.;Zhou,P.;Liu,J.J.;Yu,J.G.Phys.Chem.Chem. Phys.2014,16(38),20382.doi:10.1039/c4cp02201g
(27)Zhang,J.F.;Yu,W.L.;Liu,J.J.;Liu,B.S.Appl.Surf.Sci. 2015,358(6),457.doi:10.1016/j.apsusc.2015.08.084
(28)Guo,Z.L.;Sa,B.S.;Pathak,B.;Zhou,J.;Ahuja,R.;Sun,Z.M. Int.J.Hydrog.Energy 2014,39(5),2042.doi:10.1016/j. ijhydene.2013.11.055
(29)Yuan,J.H.;Gao,B.;Wang,W.;Wang,J.F.Acta Phys.-Chim. Sin.2015,31(7),1302.[袁俊辉,高博,汪文,王嘉赋.物理化学学报,2015,31(7),1302.]doi:10.3866/PKU. WHXB201505081
(30)Huang,C.M.;Kong,W.C.;Guan,T.P.;Wen,S.C.;Yang,T.C. Chem.Eng.Sci.2010,65(1),148.doi:10.1016/j. ces.2009.03.022
(31)Hu,X.;Hu,C.;Qu,J.Mater.Res.Bull.2008,43(11),2986. doi:10.1016/j.materresbull.2007.11.022
(32)Zhao,B.Q.;Zhang,Y.;Qiu,X.Y.;Wang,X.W.Acta Phys.Sin. 2016,65(1),014212.[赵佰强,张耘,邱晓燕,王学维.物理学报,2016,65(1),014212.]doi:10.7498/aps.65.014212
(33)Tong,H.;Ouyang,S.X.;Bi,Y.P.;Umezawa,N.;Oshikiri,M.; Ye,J.H.Adv.Mater.2012,24(2),229.doi:10.1002/ adma.201102752
(34)Nethercot,A.H.,Jr.Phys.Rev.Lett.1974,33(18),1088. doi:10.1103/PhysRevLett.33.1088
(35)Feng,C.K.;Teng,F.;Liu,Z.L.;Chang,C.;Zhao,Y.X.;Wang, S.R.;Chen,M.D.;Yao,W.Q.;Zhu,Y.F.J.Mol.Catal.AChem.2015,401,35.doi:10.1016/j.molcata.2015.02.022
(36)Segall,M.D.;Lindan,P.J.D.;Probert,M.J.;Pickard,C.J.; Hasnip,P.J.;Clark,S.J.;Payne,M.C.J.Phys.Condens. Matter.2002,14(11),2717.doi:10.1088/0953-8984/14/11/301
(37)Su,Y.C.;Xiao,L.H.;Fu,Y.C.;Zhang,P.F.;Peng,P. Sci.Sin.Phys.Mech.Astron.2011,41(1),58.[苏玉长,肖立华,伏云昌,张鹏飞,彭平.中国科学:物理学力学天文学,2011, 41(1),58.]doi:10.1360/132010-184
First-Principles Study on the Electronic and Photocatalytic Properties ofAg3XO4(X=P,As,V)
LIJiao1,2,*CHEN Zhong2
(1School of Materials Science and Engineering,Shandong University of Technology,Zibo 255049,Shandong Province,P.R.China;2School of Materials Science and Engineering,Nanyang Technological University,Singapore 639798)
In this study,the electronic structures and photocatalytic properties ofAg3XO4(X=P,As,V)were investigated using the firstprinciples based on the density functionaltheory.In comparison to Ag3PO4,Ag3VO4shows better photocatalytic stability,mainly due to the enhanced Ag―O bonds and improved Ag ion stability, butpoorer photocatalytic activity in the visible lightregion mainly due to the presence of d orbitalcharacter at the conduction band minimum(CBM)and lower valence band maximum(VBM)potentials(2.335 V,vs NHE). Ag3AsO4shows photocatalytic activity superior to Ag3PO4,which may be attributed to the following reasons:(1) the highly dispersive band structure ofthe CBM resulting from Ag s-Ag s hybridization,(2)a smaller band gap of 1.91 eV,(3)the broader absorption range and higher absorption capacity of visible light.Moreover,our theoreticalresults demonstrate thatthough Ag3XO4(X=P,As,V)species actas indirectband gap photocatalytic semiconductors,only Ag3VO4is a potentialcandidate for the photocatalytic hydrogen generation from water. The calculated results mentioned above are in good agreementwith experimentalresults.
Firstprinciples calculations;Ag3XO4(X=P,As,V);Band structure;Density ofstates; Photocatalytic property
O641
10.3866/PKU.WHXB201702085
Received:December 5,2016;Revised:January 22,2017;Published online:February 8,2017.
*Corresponding author.Email:haiyan9943@163.com;Tel:+86-533-2781317.
The projectwas supported by the College Technology Development Projectof Shandong Province,China(J15LA08).山东省高等学校科技发展计划(J15LA08)资助项目©Editorialoffice ofActa Physico-Chimica Sinica
