A tale of motor neurons and CD4+T cells: moving forward by looking back
2017-05-03AbhiramiKannanIyerKathrynJones
Abhirami Kannan Iyer, Kathryn J. Jones
Department of Anatomy and Cell Biology, Indiana University School of Medicine, Indianapolis, IN, USA
A tale of motor neurons and CD4+T cells: moving forward by looking back
Abhirami Kannan Iyer, Kathryn J. Jones*
Department of Anatomy and Cell Biology, Indiana University School of Medicine, Indianapolis, IN, USA
How to cite this article:Iyer AK, Jones KJ (2017) A tale of motor neurons and CD4+T cells: moving forward by looking back. Neural Regen Res 12(4):562-565.
Open access statement:This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.
Funding:This study was supported by grants from NIH/NINDS R01 funding NS40433.
Amyotrophic lateral sclerosis (ALS) is a fatal progressive disorder characterized by the selective degeneration of motor neurons (MN).e impact of peripheral immune status on disease progression and MN survival is becoming increasingly recognized in the ALS research fi eld. In this review, we brief l y discuss findings from mouse models of peripheral nerve injury and immunodeficiency to understand how the immune system regulates MN survival. We extend these observations to similar studies in the widely used superoxide dismutase 1 (SOD1) mouse model of ALS. Last, we present future hypotheses to identify potential causative factors that lead to immune dysregulation in ALS.e lessons from preceding work in this area offer new exciting directions to bridge the gap in our current understanding of immune-mediated neuroprotection in ALS.
amyotrophic lateral sclerosis (ALS); superoxide dismutase 1 (SOD1); immune system; SOD1 mice; motor neuron; CD4+T cells; neuroprotection
Introduction
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease characterized by loss of motor neurons (MNs) in the brainstem, motor cortex, and spinal cord. Clinically, ALS is characterized by muscular atrophy, progressive paralysis and death usually from respiratory failure within few years of diagnosis. Sporadic and familial forms of the disease share clinical and pathological hallmarks that suggest the possibility of a fi nal common pathogenic pathway leading to MN death. Mutation in the gene encoding superoxide dismutase 1 (SOD1) enzyme was the first identified genetic cause for familial ALS. Transgenic mice overexpressing the human SOD1 transgene with a causative G93A mutation (SOD1 mice, hereaer) replicate the disease course in ALS patients and have been invaluable tools to recognize the underlying pathogenesis (Moloney et al., 2014).
Parallels between Peripheral Nerve Injury and Axonal Dieback in ALS, and Role for CD4+T Cells in Both
According to one prevalent model of MN pathology in ALS, degeneration begins at the axon terminals and ascends to the cell body in a phenomenon termed ‘axonal dieback’ (Moloney et al., 2014). Facial nerve axotomy (FNA), a model for peripheral nerve injury involving transection of facial nerve axons at the stylomastoid foramen, also causes an early axonal dieback response eventually leading to MN death (Jones et al., 2015).e facial nerve injury paradigm thus replicates the earliest pathological hallmarks of human ALS disease.
FNA in recombination activating gene 2 knockout (Rag2–/–) mice, which specif i cally lack the adaptive immune system, results in significantly higher MN loss than in wild-type (WT) controls. After axotomy, MN survival is restored to WT levels in Rag2–/–recipients upon adoptive transfer of WT CD4+T cells, however, STAT6–/–CD4+T cells, that are selectively impaired in their development into the T-helper 2 (2) subset, fail to rescue MN loss (Jones et al., 2015). Although Th2 cells, specif i cally, have been shown to confer neuroprotection, axotomy leads to the development of multiple Th subsets in the draining lymph nodes of WT mice, and a predominant proinf l ammatory1 and17 cell response in SOD1 mice (Ni et al., 2016).us, it is likely that axotomy-induced pro- and anti-inflammatory Th subsets work together in the peripheral reparative process, whereas the latter are selectively recruited to the facial nucleus to warrant minimal damage to MNs.e advent ofin vivoimaging tools together with adoptive transfer of fl uorescence-expressingin vitrodifferentiated CD4+Th cells could possibly help address these questions in a direct manner.
FNA superimposed on the presymptomatic SOD1mouse defines a starting point to monitor MN loss and thereby provides a means to further understand the role of target disconnection in the SOD1 environment. In presymptomatic SOD1 mice, FNA leads to greater MN loss than in WT littermates, thus revealing an enhanced susceptibility of SOD1 MNs to peripheral nerve injury (Mariotti et al., 2002).ese studies also reveal similarities between the extent of MN loss in axotomized SOD1 and Rag2–/–mice, implicative of an underlying immunodef i ciency in SOD1 mice.e third and most important conclusion drawn from these studies is that axonal disconnection from its target musculature activates a peripheral immune response, including that of neuroprotective CD4+T cells. Surprisingly, SOD1 CD4+T cells are able to promote MN survival in axotomized Rag2–/–mice similar to their WT counterparts, suggesting an inherent neuroprotective potential within SOD1 CD4+T cells, albeit effective only when removed from the SOD1 environment (Jones et al., 2015). These findings raise the possibility that immunosuppressive factors on antigen (Ag) presenting cells (APCs) stall the neuroprotective Th2 response in these mice. Activation of T cells is initiated by recognition of Ag, but their cell function is determined by the net outcome of signaling cascades that are activated upon binding of co-stimulatory or co-inhibitory receptors on T cells to their cognate ligands on APCs.us, it is possible that SOD1 CD4+T cells are dysfunctional in their endogenous environment due to either a reduced expression of co-stimulatory ligands or enhanced expression of co-inhibitory ligands on APCs. Currently studies are underway in our laboratory to address this hypothesis with an ultimate aim to identify targets that can be selectively blocked or enhanced to stimulate the neuroprotective CD4+Th2 responses.
Dissecting Major Histocompatibility Complex (MHC) Class II-Mediated Ag Presentation in Peripheral Nerve Injury and ALS-Like SOD1 Mice - Neighborhood Matters for CD4+T Cell Function
differentiation of a naïve CD4+T cell into ef f ector subsets is instructed by the dose of Ag, the type of APC displaying Ag-MHC class II complexes, and the length of stimulation period, with a lower antigenic dose in a prolonged stimulation creating a Th2 skewing microenvironment (Rothoeet al., 2003). It has long been identified that Ags triggering CD4+T cell reactivity in autoimmune multiple sclerosis are myelin proteins; however, the source of Ag triggering peripheral immune activation in facial nerve injury or ALS are yet unknown. In spinal cord injury, the neuroprotective versus neuropathological effects of autoimmune CD4+T cells are debatable, but immunizing mice with myelin basic protein prior to FNA intensified MN loss, thereby negating a neuroprotective role for autoimmune CD4+T cells under such conditions (Ankeny and Popovich, 2007). Although yet to be confirmed, it is very likely that debris forming at the neuromuscular junction (NMJ) due to target disconnection could be the Ag source that elicits the MN-protective Th2 response. If this is indeed the case, further analysis to unravel the neuronal versus muscular origin of such an Ag could have implications in designing vaccines that can selectively boost neuroprotective immune responses in ALS patients.
Robust activation of resident glial cells has been reported in the facial nucleus following peripheral nerve injury (Quan and Gao, 2006), supporting the idea that glia could present Ag and provide cues to CD4+T cells centrally. Studies utilizing bone marrow chimeric mice that harbor selective deletion of MHC class II on APCs in either the peripheral or central compartment, followed by adoptive transfer of CD4+T cells and FNA, established that MHC class II-mediated Ag presentation in both the compartments is indispensable for CD4+T cells to confer neuroprotection. Thus, upon axotomy, peripheral MHC class II+APCs provide the initial Ag-specif i c stimulus to CD4+T cells, which are then reactivated by centrally-residing MHC class II+APCs (Jones et al., 2015). Further, there is evidence for CD4+T cell infiltration within the facial motor nucleus between days 7 and 21 aer axotomy (Raivich et al., 1998).us, there appears to be a requisite for central Ag presentation by MHC class II in order to confer specif i city to inf i ltrating CD4+T cells and sustain their responseviadisplay of the same activating Ags that induced them in the periphery.
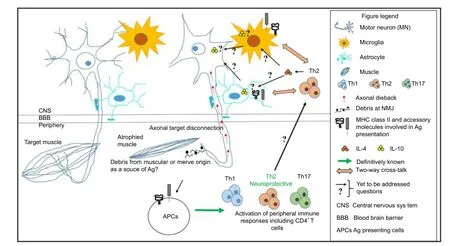
Figure 1 Neuro-immune interactions involved in motor neuron (MN) survival, lessons learned and moving forward.
Transgenic mice generated to express SOD1 in selective tissues show MN degeneration only when mutant protein is expressed in both neurons and microglia or astrocytes, the CNS resident immune effector cells, suggesting a pathogenic role for multiple cell types in disease onset and progression (Moloney et al., 2014). In line with these observations, laser microdissection studies in our laboratory have revealed that axotomy in WT mice induces a gene expression pattern that is pro-regenerative within MNs and pro-inflammatory in the surrounding neuropil (Jones et al., 2015). Additional reconstitution experiments of WT CD4+T cells into Rag2–/–mice have shown that MN survival is mediated by an initial wave of microglial and astroglial activation that is tightly regulated and declines around 28 days post axotomy (unpublished data, Setter DO, Jones KJ). On the contrary, SOD1 mice show reduced astrocyte activation and constitutive expression of proinflammatory TNF-α that fails to resolve at later time-points aer axotomy (Mesnard et al., 2011). An independent study by another group has shown that microglia isolated from the spinal cords of SOD1 mice adopt a peripheral dendritic cell-like phenotype, with concurrent upregulation of T cell co-stimulatory and adhesion molecules such as CD86 and CD54, respectively (Chiu et al., 2008). Collectively, these studies are indicative of microglial and astrocytic activation upon axonal disconnection from its target musculature, however, it is unclear which resident glial cell upregulates MHC class II expression for central Ag presentation.erefore, more direct approaches such as the Cre/lox technology to selectively delete MHC class II within microglia or astrocytes, are required to dissect the role of each glial cell in directing the CD4+T cell responses to axonal dieback. Additionally, a detailed temporal and cellular characterization examining the expression of MHC class II and accessory molecules (namely, co-stimulatory/co-inhibitory molecules, cytokines and chemokines) on APCs together with their cognate receptors on CD4+T cells is required in both the periphery and the brain. Although upregulation of immune markers such as MHC class II and co-stimulatory molecules has been described in the spinal cords of human ALS patients and SOD1 mice (Chiu et al., 2008), the nature of initial immune response to axonal target disconnection, reported to occur prior to MN loss at postnatal day 40 in SOD1 mice (Moloney et al., 2014) is currently unknown.us, studies incorporating earlier time-points are necessary to capture the primary immune response to axonal dieback.
Concluding Remarks
Activation of the innate and adaptive immune system is becoming increasingly witnessed in neurodegenerative diseases such as ALS. At a time when CD4+T cells are perceived as pathological drivers in neurodegenerative diseases, recent insights into their neuroprotective role in ALS, provides a refreshing take on the functional capabilities of CD4+T cells. While we have come a long way in understanding ALS pathogenesis, we have only scratched the surface in understanding the neuro-immune interactions involved in facilitating MN survival. As summarized in Figure 1, the source of Ag triggering such immune responses, the cellular source, kinetics of MHC class II and accessory molecules involved in Ag presentation in both central and peripheral compartments, and the mechanism(s) by which Th2 cells support MN survival, are some of the great unknowns that currently blur our understanding of this immune-mediated neuroprotection. Research in this area will pave the way for discovery of easily accessible biomarkers in ALS to assist in early diagnosis, disease monitoring, and assessing therapeutic benef i ts. A combinatorial approach aimed at preserving the neuromuscular junction and inducing the neuroprotective immune response appears to be the way forward in developing therapies for this multifactorial disease.
Acknowledgements:We would like to thank Elizabeth M. Runge for critical reading and comments on this article. Also, we apologize to authors whose work could not be cited here due to article space constraints. This study was supported by grants from NIH/NINDS R01 funding NS40433.
Ankeny DP, Popovich PG (2007) Central nervous system and non-central nervous system antigen vaccines exacerbate neuropathology caused by nerve injury. Eur J Neurosci 25:2053-2064.
Chiu IM, Chen A, Zheng Y, Kosaras B, Tsisoglou SA, Vartanian TK, Brown RH, Jr., Carroll MC (2008) T lymphocytes potentiate endogenous neuroprotective inf l ammation in a mouse model of ALS. Proc Natl Acad Sci U S A 105:17913-17918.
Jones KJ, Lovett-Racke AE, Walker CL, Sanders VM (2015) CD4+T Cells and Neuroprotection: Relevance to Motoneuron Injury and Disease. J Neuroimmune Pharmacol 10:587-594.
Mariotti R, Cristino L, Bressan C, Boscolo S, Bentivoglio M (2002) Altered reaction of facial motoneurons to axonal damage in the presymptomatic phase of a murine model of familial amyotrophic lateral sclerosis. Neuroscience 115:331-335.
Mesnard NA, Sanders VM, Jones KJ (2011) Differential gene expression in the axotomized facial motor nucleus of presymptomatic SOD1 mice. J Comp Neurol 519:3488-3506.
Moloney EB, de Winter F, Verhaagen J (2014) ALS as a distal axonopathy: molecular mechanisms affecting neuromuscular junction stability in the presymptomatic stages of the disease. Front Neurosci 8:252.
Ni A, Yang T, Mesnard-Hoaglin NA, Gutierrez R, Stubbs EB, Jr., McGuire SO, Sanders VM, Jones KJ, Foecking EM, Xin J (2016) Th17 cell response in sod1g93a mice following motor nerve injury. Mediators Inf l amm 2016:6131234.
Quan SM, ZQ Gao (2006) Immunobiology of facial nerve repair and regeneration. J Otol 1:107-115.
Raivich G, Jones LL, Kloss CU, Werner A, Neumann H, Kreutzberg GW (1998) Immune surveillance in the injured nervous system: T-lymphocytes invade the axotomized mouse facial motor nucleus and aggregate around sites of neuronal degeneration. J Neurosci 18:5804-5816.
*< class="emphasis_italic">Correspondence to: Kathryn J. Jones, Ph.D., kjjones1@iupui.edu.
Kathryn J. Jones, Ph.D., kjjones1@iupui.edu.
orcid: 0000-0002-0580-5436 (Kathryn J. Jones)
10.4103/1673-5374.205086
Accepted: 2017-02-28
杂志排行

中国神经再生研究(英文版)的其它文章
- Recovery of multiply injured ascending reticular activating systems in a stroke patient
- Neuroprotective mechanism of Kai Xin San: upregulation of hippocampal insulin-degrading enzyme protein expression and acceleration of amyloid-beta degradation
- Mitomycin C induces apoptosis in human epidural scar fi broblasts after surgical decompression for spinal cord injury
- Exenatide promotes regeneration of injured rat sciatic nerve
- Recombinant human fi broblast growth factor-2 promotes nerve regeneration and functional recovery after mental nerve crush injury
- Ca2+involvement in activation of extracellular-signalregulated-kinase 1/2 and m-calpain after axotomy of the sciatic nerve