The complexities underlying age-related macular degeneration: could amyloid beta play an important role?
2017-05-03savannahLynnEloiseKeelingrosieMundayGagandeepGabhahelenGriffithsAndrewLoteryArjunaratnayaka
savannah A. Lynn, Eloise Keeling, rosie Munday, Gagandeep Gabha, helen Griffiths, Andrew J. Lotery,, J. Arjuna ratnayaka,
1 Clinical and Experimental Sciences, Faculty of Medicine, University of Southampton, Southampton, United Kingdom
2 Eye Unit, University Southampton NHS Trust, Southampton, United Kingdom
The complexities underlying age-related macular degeneration: could amyloid beta play an important role?
savannah A. Lynn1, Eloise Keeling1, rosie Munday1, Gagandeep Gabha1, helen Griffiths1, Andrew J. Lotery1,2, J. Arjuna ratnayaka1,*
1 Clinical and Experimental Sciences, Faculty of Medicine, University of Southampton, Southampton, United Kingdom
2 Eye Unit, University Southampton NHS Trust, Southampton, United Kingdom
How to cite this article:Lynn SA, Keeling E, Munday R, Gabha G, Grif fi ths H, Lotery AJ, Ratnayaka JA (2017) Te complexities underlying age-related macular degeneration: could amyloid beta play an important role? Neural Regen Res 12(4):538-548.
Open access statement:Tis is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.
Funding:Tis work was funded by the National Centre for the Replacement Refnement & Reduction of Animals in Research (NC3R: Grant # NC/L001152/1), the Macular Society, UK, National Eye Research Centre, and the Gift of Sight Appeal.
Age-related macular degeneration (AMD) causes irreversible loss of central vision for which there is no effective treatment. Incipient pathology is thought to occur in the retina for many years before AMD manifests from midlife onwards to affect a large proportion of the elderly. Although genetic as well as non-genetic/environmental risks are recognized, its complex aetiology makes it dif fi cult to identify susceptibility, or indeed what type of AMD develops or how quickly it progresses in different individuals. Here we summarize the literature describing how the Alzheimer’s-linked amyloid beta (Aβ) group of misfolding proteins accumulate in the retina.e discovery of this key driver of Alzheimer’s disease in the senescent retina was unexpected and surprising, enabling an altogether different perspective of AMD. We argue that Aβ fundamentally differs from other substances which accumulate in the ageing retina, and discuss our latest fi ndings from a mouse model in which physiological amounts of Aβ were subretinally-injected to recapitulate salient features of early AMD within a short period. Our discoveries as well as those of others suggest the pattern of Aβ accumulation and pathology in donor aged/AMD tissues are closely reproduced in mice, including late-stage AMD phenotypes, which makes them highly attractive to study dynamic aspects of Aβ-mediated retinopathy. Furthermore, we discuss our fi ndings revealing how Aβ behaves at single-cell resolution, and consider the long-term implications for neuroretinal function. We propose Aβ as a key element in switching to a diseased retinal phenotype, which is now being used as a biomarker for latestage AMD.
amyloid beta (Aβ); retinal neurons; retina; mouse models; age related macular degeneration (AMD)
Introduction
As the name implies, AMD is characterized by irreversible damage to the central region of the retina, termed the macula (Figure 1B), which provides focused vision. In most cases, primary pathology develops in the RPE, which has therefore been a major focus of research (Marmor and Wolfensberger, 1998; Bhutto and Lutty, 2012; Khandhadia et al., 2012). Incipient retinal pathology is thought to develop at cellular level for many years although patients do not initially report any visual problems. The development of protein/ lipid aggregates at the RPE-BrM interface in the macula is considered to be the fi rst clinical indicator of increased susceptibility (Sarks et al., 1999). Such deposits, termed drusen, appear as yellow-white spots when viewed through an ophthalmoscope (Figure 1C), although even at this stage patients remain largely asymptomatic (Bhutto and Lutty, 2012; Khandhadia et al., 2012). Early AMD was observed in 3.8% of individuals as young as 35–45 years of age (Korb et al., 2014), which rose to 9.8% in those aged 65 years or older (Klein et al., 2010).is early stage may then progress to intermediate and late AMD, where two broadly-defined phenotypes develop: geographic atrophy (dry AMD or GA) or vascular (wet) AMD (Lotery and Trump, 2007; Khandhadia et al., 2012). By the time individuals reach the 8thdecade of life, the prevalence of sight threatening latestage AMD has increased to ~12% (Friedman et al., 2004). Of the two late-stage phenotypes, GA AMD is defined by the development of a macular lesion, corresponding to an area in which RPE and photoreceptors have atrophied. As a consequence, the underlying choroid becomes more visible when viewed through an ophthalmoscope (Figure 1D). Vascular AMD is characterized by primary RPE pathology and a compromised BRB through which proliferative choroidal vessels invade the retinal environment. These leaky vessels release fluids or exudates which accumulate beneath the RPE/retina to cause retinal detachment, and form a fi brous scar over time (Figure 1E). Vascular AMD is routinely managedviaintravitreal injections of anti-vascular endothelial growth factor (VEGF) inhibitors such as ranibizumab (Lucentis) and aflibercept (Eylea) or the of f-label bevacizumab (Avastin) (Amoaku et al., 2015). Although patients report signif i cant sight improvements, this strategy is largely unsatisfactory in the long term as initial visual gains are almost impossible to maintain, whilst some individuals do not respond to treatments at all. Critically, prolonged treatment appears to cause a switch in some patients to the GA form of AMD (Lois et al., 2013; Grunwald et al., 2015) which has no treatment at all. It appears that relative frequencies of GA and vascular AMD are broadly similar if only late disease is compared (Owen et al., 2012), which means that at present, approximately half of late AMD patients do not even have a strategy to manage the disease. This lack of meaningful AMD treatment bodes poorly for aging populations throughout the world. For instance, AMD currently af f ects > half a million individuals in the United Kingdom alone, and is expected to increase to 680,000 cases by 2020 (source Macular Society, UK). Globally, early stages of AMD are estimated to af f ect > 150 million individuals (Wong et al., 2014). As populations age, an increasing number of AMD cases are also being reported in rapidly developing countries (Krishnan et al., 2010; Ye et al., 2014; Elias et al., 2015) which add to existing high AMD numbers in the developed world.e disease is also aggressive, rapidly progressing from largely asymptomatic early stages to advanced AMD which is when sight loss occurs. For instance, ~15% of individuals with soindistinct drusen or 20% of those with RPE abnormalities progressed to latestage AMD within a 10-year period (Klein et al., 2002).is rate of disease progression is ref l ected in the high incidence of late-stage AMD patients, estimated to be approximately 71,000 newly diagnosed cases that are annually recorded in the United Kingdom (Owen et al., 2012).e high prevalence of advanced AMD cases is also evident worldwide, which is predicted to affect approximately 10 million individuals (Wong et al., 2014).
It is unclear whether the two different AMD phenotypes result from the consequences of a single disease affecting different tissues of the outer retina or is in fact a combination of several diseases. The conventional view has been that primary RPE impairment leads to subsequent photoreceptor damage and eventual sight loss (Lotery and Trump, 2007; Khandhadia et al., 2012). It must be noted however that the primary site of AMD pathology is not solely restricted to the RPE, but can also manifest in the photoreceptor layer, as well as the choroid (Spaide, 2009; Bird et al., 2014). For instance, in some cases photoreceptor atrophy is observed in the absence of any underlying RPE pathology (Bird et al., 2014). differences in where disease fi rst manifests make it dif ficult to study its aetiology, which is made even more challenging as AMD diverges to distinct GA and vascular phenotypes. To complicate matters further, in some instances AMD can also develop in the absence of any macular drusen. Such observations reinforce the idea that complex and ill-def i ned mechanisms may be at play which trigger and/or drive AMD at different speeds towards different outcomes in different individuals. The Age Related Eye Disease Study (AREDS) classif i es and grades AMD to ref l ect these subtleties and to account for the diverse range of reported clinical phenotypes (Davis et al., 2005; Ferris et al., 2005). Recently, the appearance of drusen between the RPE and photoreceptors, referred to as reticular pseudodrusen, was shown to indicate a greater risk of progressing to advanced AMD compared to eyes with drusen only (Spaide, 2013; Sivaprasad et al., 2016). However, classif i cation is still largely based on clinical observations, which means that incipient cellular changes that occur before macular drusen becomes apparent may remain undetected. Recent evidence from patients treated with anti-VEGF inhibitors could potentially offer clues to the underlying pathology of AMD, as prolonged treatment can induce GA. The mechanisms by which RPE damage occurs under these circumstances are still unclear, although a putative correlation was observed between increased numbers of anti-VEGF injections and the severity of RPE damage (Lois et al., 2013). One suggestion is that this may in fact have unmasked GA to be the actual AMD phenotype. Further work is of course required to investigate this intriguing hypothesis.
Efects of Genetic and Non-Genetic Risk Factors in a Changing Retinal Landscape
AMD is driven by rare as well as common genetic risk factors. Our work has contributed signif i cantly to revealing its genetic architecture (Fritsche et al., 2013, 2015), including the roles of fi brillin (Ratnapriya et al., 2014), fi bulin 5 (Stone et al., 2004), complement proteins and their regulators (Ennis et al., 2008; Gibson et al., 2012; Sofat et al., 2012), ApoE (McKay et al., 2011) and HLA (Goverdhan et al., 2005), as well as the roles of genes in chromosome 6p21.3 (Cipriani et al., 2012). Our findings have also provided molecular insights into how a variant cystatin C associated with AMD could impair the RPE (Paraoan et al., 2004; Ratnayaka et al., 2007b) and how extracellular proteins such as SPARC alter the RPE-BrM interface (Ratnayaka et al., 2007a). Thus far however, the molecular/biochemical consequences of most risk genes and how they might alter tissues in the outer retina remains to be determined. AMD is also driven by non-genetic or environmental risk factors such as nutrition and smoking (Chew et al., 2013; Chiu et al., 2014; Woodell and Rohrer, 2014), which play significant roles in predisposing the senescent retina to disease. Hence, as conditions in the retina gradually alter with age, different risk factors exert their influence singly and in concert to bring about irreversible change. How precisely these factors inf l uence the development of retinopathy, and to what extent, are as yet unclear and may largely depend on the individuals’ genetic/non-genetic backgrounds. For instance, the deposition of cholesterol in the retina is associated with developing AMD (Rudolf and Curcio, 2009; Klein et al., 2010). Several cholesterol-linked genes such as CETP, ABCA1, LIPC and ApoE have also been correlated with AMD, but not necessarily the amounts of cholesterol deposited in retinas of individuals carrying mutations or isoforms of these genes. Hence, genetic influences on how much retinal cholesterol becomes deposited may be affected further by an individuals’ diet, the presence of other proteins as well as cholesterol clearance/turnover mechanisms. The formation/deposition of advanced glycation end products (AGEs) in the senescent retina also represents a further shitowards disease (Glenn and Stitt, 2009). Other factors such as Zn2+and Fe3+that accumulate in the RPE-BrM interface, for which there appears to be no obvious genetic link (Hahn et al., 2003; Lengyel et al., 2007), further contributes to alter the biophysical landscape of the retina.e accumulation of amyloid beta (Aβ) in the aging retina represents another such risk factor, and the focus of this review.
Aβ is a family of misfolding proteins correlated with neurodegenerative diseases such as AD. Aβ is generated from sequential cleavage of the ubiquitously expressed amyloid precursor protein (APP) and is secreted extracellularly by neurons (Jarrett et al., 1993; Hardy and Selkoe, 2002). Given its central role in AD, histological data showing high quantities of Aβ persistently accumulating in aged/AMD retinas were somewhat unexpected, particularly since a genetic correlation between Aβ and AMD is yet to be established. It is also possible that any genetic correlation is either weak or non-existent, and that Aβ accumulation is akin to the deposition of other substances such as AGEs, Zn2+and Fe3+, lipofuscin or cholesterol in aged/AMD retinas. Why this highly neurotoxic group of proteins consistently aggregate in the aging retina, and to what extent they contribute to AMD, remains to be fully addressed. Our interest also stems from the fact that Aβ is fundamentally different to many of the aforementioned substances or compounds accumulating in aged retinas, as Aβ appear to have the capacity to trigger/drive multiple disease pathways. For example, Aβ is associated with inducing neuronal senescence (He et al., 2013), disrupting cell membrane integrity (Williams et al., 2010), altering neuronal long-term potentiation (Townsend et al., 2006), inducing synaptic loss (Lacor et al., 2007; Shankar et al., 2007) as well as inducing memory defects in animal models (Duf f et al., 1996; Citron et al., 1997) amongst others. Our earlier work also revealed a pathway through which Aβ causes neuronal death (Soura et al., 2012). Additionally, Aβ has the capacity to activate both the alternative and classical complement pathways (Bradt et al., 1998), elicit neuroinf l ammation (Minter et al., 2016) as well as induce vascular permeability and angiogenesis (Jef f eries et al., 2013). We consider this to be a key feature by which Aβ potentially differs from other substances that aggregate in the senescent retina.
Aβ Synthesis and Assembly- Insights into Aβ Accumulation in the Senescent Retina
differences in the way APP is cleaved yields a variety of Aβ peptides of which Aβ1–40and Aβ1–42species are the most common (Benilova et al., 2012). Aβ levels as well as the relative ratios of Aβ1–40vs. Aβ1–42are altered in AD patients (Hardy and Selkoe, 2002; Kuperstein et al., 2010). Moreover, Aβ peptides may be modified by further enzymatic activity resulting in a mixture of > 20 different Aβ peptides.e process of Aβ aggregation is described by the nucleation-dependent polymerisation model in which monomeric Aβ assembles into dimers, trimers and oligomers followed by a switch to a faster phase where larger structures including fibrillar Aβ are formed (Jarrett et al., 1993; Ward et al., 2000; Lee et al., 2011).e formation of smaller soluble Aβ forms as well as larger insoluble Aβ aggregates may be observedin vitro(Bitan et al., 2005; Benilova et al., 2012). Importantly, these Aβ forms can also be isolated from AD brains (Hardy and Selkoe, 2002; Shankar et al., 2008; Noguchi et al., 2009). Oligomeric Aβ has been shown to have a closer relationship with AD progression (McLean et al., 1999; Mc Donald et al., 2010) compared to Aβ plaques which do not necessarily correlate with disease severity (Perrin et al., 2009).e potency of oligomeric Aβ is illustrated by experiments showing how its ability to penetrate biological membranes diminishes as oligomers assemble to a fi brillar state (Williams et al., 2010). A compelling case can therefore be made for oligomeric Aβ as key driver of degeneration, further supported by a plethora of new fi ndings claiming the identif i cation of various toxic Aβ oligomers. Amyloid plaqueshave been proposed to exist in a dynamic equilibrium with oligomeric Aβ resulting in a local spillover of neurotoxic Aβ species in adjacent tissues (Tseng et al., 1999; DeMattos et al., 2002). Oligomeric Aβ may also mediate ef f ects at some distances from established plaques (Benilova et al., 2012) with the latter perhaps acting as a reservoir for toxic Aβ species. For these reasons, we routinely utilizein vitropreparations of oligomeric Aβ in our experiments. Of the diverse Aβ species, Aβ1–42in particular has garnered considerable attention, as the addition of a hydrophobic isoleucine and alanine residue at its C-terminus is thought to facilitate rapid aggregation compared to Aβ1–40(Kim and Hecht, 2005). Aβ1–42is also thought to provide a nucleus for subsequent fibril formation (Jarrett et al., 1993) and is associated with a plethora of neurodegenerative events (Selkoe, 2008; Benilova et al., 2012). For these reasons we and many others utilize oligomeric Aβ1–42in our experiments, although this may exclude putative ef f ects of an as yet unidentified ‘toxic Aβ species’ or indeed a ‘toxic Aβ oligomeric soup’ consisting of several Aβ species, which might exist in AD brains (Benilova et al., 2012). We employ a method ofin vitroAβ preparation used by Broersen and colleagues (Broersen et al., 2011) in which we are able to clearly visualise the progressive assembly of smaller, soluble Aβ forms (Soura et al., 2012). The resulting Aβ not only conforms to the expected size of Aβ oligomers, but their authenticity has also been confirmed by utilizing antibodies that specifically detect Aβ (Taylor-Walker et al., 2016). Concerns have been raised about the use of non-physiological Aβ concentrations in experiments that range from levels in excess of 1 μM Aβ to as high as 10–40 μM Aβ. Researchers are however driven to these extremes due to issues including (a) requirements for high Aβ concentrations which may be necessary for monomeric Aβ to assemble into toxic oligomers, (b) attempts to recapitulate disease conditions in a matter of several hours or days that would otherwise require several decades, as well as (c) the fact thatin vitrogenerated Aβ appear to be less toxic compared to Aβ oligomers generated from cultured cells or brain tissues (Selkoe, 2008; Benilova et al., 2012). In our view, acute dosage using non-physiological Aβ concentrations does not necessarily replicate chronic Aβ exposure to which cells/tissues of the ageing retina are subjected to over many years. Hence, our studies use physiological Aβ concentrations in the picomolar to nanomolar levels, and only as high as 1 μM Aβ, which may be considered to realistically recapitulate chronic Aβ exposure in native tissues. In reality however, the extent of Aβ exposure in different tissues may vary considerably, and may even differ between groups of cells in a given tissue as conditions such as temperature, pH, the presence of metals and other proteins/lipids influence the speed of aggregation, as well as the availability of toxic Aβ oligomers in a given locality (Atwood et al., 2003; Benilova et al., 2012). Hence, the experimenter can only hope to inf l uence Aβ assembly underin vitroconditions, as once introduced into a biological system, the likelihood of controlling Aβ becomes largely academic. Toxic Aβ oligomers could therefore rapidly form and/or persist in a given micro-environment, whilst their assembly may follow an alternative fate in another. In this respect, cell cultures may be more amenable to manipulating Aβ, as this becomes nearly impossible underin vivoconditions.
What is the Role of Aβ in the Senescent Retina?
Histological data from human donor eyes and mouse tissues reveal specif i c locations where Aβ accumulates in the outer retina.e focus of Aβ deposition appears to be in the RPE and BrM, although it also aggregates in photoreceptor outer segments (POS) and within choroidal vessels (Hoh et al., 2010; Ohno-Matsui, 2011; Ratnayaka et al., 2015). Histological analysis has also revealed Aβ within drusen organised into structures between 0.25–20 μm in diameter termed amyloid vesicles (Anderson et al., 2004; Luibl et al., 2006; Isas et al., 2010).e interior of these vesicles were organised into concentric ring-like layers with different electron densities and permeated with Aβ immunoreactivity (Anderson et al., 2004). Drusen were found to contain one or more such amyloid vesicles, which constituted a significant portion of their volume.e authors suggest that multiple amyloid cores could indicate that these drusen may have coalesced from several smaller drusen (Anderson et al., 2004), as is indeed the case since drusen is known to alter shape and change over time (Sarks et al., 1999). Furthermore, their fi ndings showed Aβ to be in retinas with moderate to high amounts of drusen, suggesting that Aβ might also be associated with progressive stages of AMD (Anderson et al., 2004).e presence of Aβ within drusen has also been confi rmed histologically by several other groups (Mullins et al., 2000; Johnson et al., 2002; Dentchev et al., 2003; Luibl et al., 2006; Isas et al., 2010). Use of antibodies which recognised distinct Aβ structures indicated oligomeric rather than monomeric or fi brillar Aβ to form the core of drusen, in close proximity to the inner collagenous layer of BrM (Luibl et al., 2006; Isas et al., 2010) and the site of drusen biogenesis (Rudolf et al., 2008). Moreover, oligomeric Aβ appeared to form the majority of Aβ within drusen (Luibl et al., 2006; Isas et al., 2010). In contrast, fi brillar Aβ was found to be predominantly localised to the periphery of amyloid vesicles (Johnson et al., 2002; Anderson et al., 2004; Luibl et al., 2006; Isas et al., 2010). Assembly of Aβ as senile plaques in AD brains is considered to act as a scaf f old onto which components such as ApoE, complement proteins, immunoglobulins, fibroblast growth factor, AGE products, as well as prion proteins amongst others subsequently bind (Armstrong, 2009). Aβ also has the propensity to bind metals such as Zn2+and Fe3+(Atwood et al., 2003), whilst ef f ects of post-translational modif i cation and cross-linking has been shown to decrease its solubility and increase resistance to proteases (Smith et al., 1994).ese events may signif i cantly alter the biophysical properties of senile plaques over long periods. A similar process might also occur in the senescent retina, in which oligomeric Aβ forms a scaf f oldin proximity to the inner collagenous layer of BrM, around which drusen constituents subsequently accumulate. This possibility is consistent with the hypothesis that chronic local inflammatory and immune-mediated events at the RPE-BrM interface play critical roles in drusen formation and hence in the development of AMD (Anderson et al., 2010). Ample evidence for complement proteins interacting with amyloid vesicles have also been reported by immunofluorescence studies in fixed donor AMD tissues (Johnson et al., 2002). Other types of interactions between drusen components and Aβ have also become evident. For instance, the exposure of RPE cells to iron was shown to upregulate APP and increase Aβ synthesis (Guo et al., 2014), whilst senescent C57BL/6 wildtype mice fed a high cholesterol diet developed sub-RPE Aβ deposits (Wang et al., 2012a).ere is also strong evidence to suggest that Aβ ef f ects are not limited to early AMD but may also manifest in late stages. For instance, exposure of cultured RPE to high Aβ levels resulted in elevated secretion of pro-angiogenic VEGF, whilst human umbilical vein endothelial cells spontaneously formed tubes when exposed to conditioned media from Aβ-treated RPE (Yoshida et al., 2005). Furthermore, injection of Aβ into zebraf i sh eyes resulted in rapid proliferation of retinal capillaries (Cunvong et al., 2013). Aβ treatment of cultured RPE also induced cellular senescence and created a pro-inf l ammatory microenvironment (Cao et al., 2013). Collectively, this evidence suggests that Aβ could play an important role in progressive stages of AMD. Much like a spider at the centre of a complex web, Aβ appears to have the capacity to trigger and/ or drive multiple disease pathways associated with early as well as late-stage AMD.
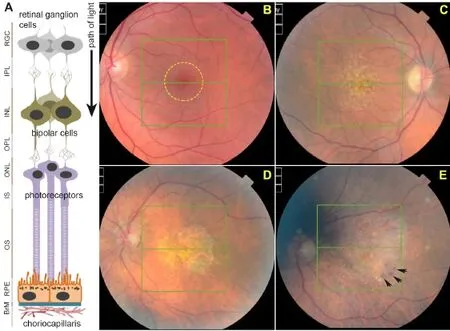
Figure 1 Structure of the retina and development of age-related macular degeneration (AMD) pathology.
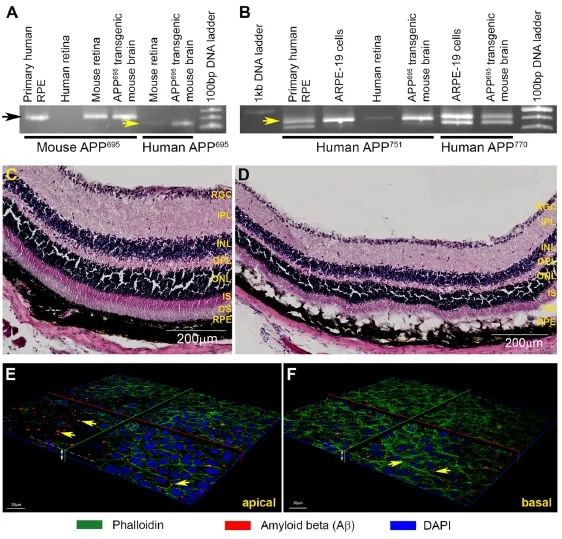
Figure 2 Efects of amyloid beta (Aβ) in living mouse retinas and in cultured retinal neurons.
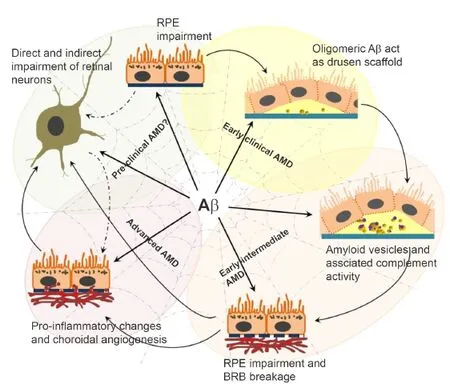
Figure 3 Amyloid beta (Aβ) has the capacity to trigger/drive multiple disease mechanisms in the senescent retina.
Although fi xed eye tissues from aged/AMD donors provide unequivocal histological insights into the importance of Aβ in retinal degeneration, they are largely unsuitable for studies investigating more dynamic aspects of Aβ-mediated retinal pathophysiology. Fortunately, we are able to call upon the aid of rodent models, as their pattern of retinal Aβ aggregation is similar to that observed in donor human eyes. For instance, Aβ has been shown to accumulate in the RPEBrM interface (Hoh et al., 2010; Wang et al., 2012a), POS and in retinal and choroidal vessels of wildtype C57BL/6 mice (Hoh et al., 2010), as well as in the basal side of RPE and in the choroid of Long-Evans rats (Zhao et al., 2015). Aβ also accumulates in sub-RPE deposits and choroidal neovascular lesions in ApoE4-HFC transgenic mice (Malek et al., 2005; Ding et al., 2008), in outer and inner plexiform retinal layers of APPswe/PS1ΔE9 transgenic mice (Perez et al., 2009), as well as in the retinal ganglion cell (RGC) and inner nuclear layers of Tg2576 transgenic mice (Dutescu et al., 2009).ese fi ndings demonstrate that mice represent a powerful model with which Aβ-mediated retinal pathology could be studied under realisticin vivoconditions. To take advantage of this, we developed a murine model where a specif i c type (oligomeric), species (Aβ1–42) and Aβ quantity (nanomolar) was introduced into living retinas in a controlled manner. In the past, others have introduced Aβ intothe retinal environment by means of an intravitreal injection (Walsh et al., 2002, 2005; Howlett et al., 2011). However, we reasoned that introduction of Aβviaa subretinal route would recapitulate conditions in the senescent retina and associated tissues more accurately, particularly as the RPE is considered to be the major source of Aβ synthesis/secretion in the posterior eye (Ohno-Matsui, 2011; Ratnayaka et al., 2015).e importance of RPE for Aβ synthesis has been conf i rmed by immunohistochemistry which showed the expression of APP and the Aβ-producing enzyme β-secretase in these cells (Yoshida et al., 2005), and by cell culture and ELISA methods which showed that isolated mouse RPE secreted higher Aβ levels compared to cells from younger animals (Wang et al., 2012b). As described earlier, histological evidence from donor AMD tissues as well as wildtype and transgenic mice also conf i rmed the importance of the RPE for retinal Aβ production. Our studies showed the expression of the three alternatively-spliced mRNA APP isoforms; APP695, APP751and APP770(Ponte et al., 1988; Tanzi et al., 1988) in retinal and RPE homogenates (Figure 2A, B), revealing the breadth of APP expression in tissues of the outer retina (Lynn et al., 2016). For our mouse experiments, we used nM Aβ quantities, in contrast to doses as high as 40 μM Aβ used intravitreally in the past (Walsh et al., 2002; Anderson et al., 2009; Fisichella et al., 2016). We also used the highly toxic oligomeric Aβ instead of fibrillar Aβ used by others (Howlett et al., 2011) which is considered to be less pathogenic. We utilized wildtype C57BL/6 mice as their dark retinal pigmentation allowed better visualisation of incipient as well as advanced stages of retinal pathology. One week after subretinal Aβ injection, mice displayed normal retinal functions compared to vehicle injected controls as measured by full-f i eld electroretinogram (ERGs).is was reassuring since it indicated a functional living retina unimpaired by surgical procedures or gross retinal pathology. Subretinal Aβ was introduced in a manner such that only half of the retina became exposed, whilst the majority of the retina remained unexposed and therefore acted as an internal control. Furthermore, the overall area of Aβ exposure was relatively small, typically 20-30% of the total retina, which recapitulated the likeness of localised retinal pathology that develops in the macula of AMD patients (Lynn et al., 2016). We note that early changes in AMD patients are similarly undetectable by fullfi eld ERGs (de Oliveira Dias et al., 2016). However, whilst full- fi eld ERGs detected no changes, immunohistochemical analysis revealed a very different picture (Figure 2C, D). We observed pigment abnormalitiesviafunduscopy which were correlated with hyperplastic RPE and disrupted RPEBrM; indicative of a compromised BRB. Mice also showed loss of POS (Lynn et al., 2016; Ratnayaka and Lynn, 2016), which collectively constituted salient features of early-intermediate AMD (Davis et al., 2005; Khandhadia et al., 2012). Comparable histopathological features including the upregulation of interleukins 6 and 8 in the RPE-choroid were observed by colleagues using a similarin vivomodel (Liu et al., 2015). We are therefore con fi dent that our acute Aβ-injection mouse model mimics subtle as well as specif i c features of AMD.
We next sought to determine specif i c locations in which Aβ could potentially aggregate in retinas of these mice. One week aer injection, Aβ was observed in the RPE-BrM interface, on POS as well as in the outer plexiform layer, inner nuclear layer and RGC (Lynn et al., 2016; Ratnayaka and Lynn, 2016; Taylor-Walker et al., 2016).is pattern of Aβ aggregation showed a striking correlation with areas of pathology indicated by parallel histological studies. Although pathology of the inner retina is not associated with AMDper se, areas of Aβ aggregation in the retina corresponded to neurons/cell-layers known to be immunopositive for APP and/or Aβ (Guo et al., 2007; Dutescu et al., 2009; Hoh et al., 2010; Kipfer-Kauer et al., 2010; Dasari et al., 2011; Koronyo-Hamaoui et al., 2011; Wang et al., 2011). We found it surprising that exogenously delivered human Aβ1–42should also colocalize to these specif i c retinal locations. We wanted to establish whether this implied that certain retinal layers or types of retinal neurons were in some way susceptible to Aβ. To study this at single cell-resolution we turned to cultured retinal neurons, as suchin vitroapproaches provide the necessary control and allows use of powerful microscopes to address this question. Chronic exposure to 1 μM Aβ1–42resulted in rapid internalisation of oligomeric Aβ which remained sequestered within neurons for several days, whilst larger Aβ forms appeared to aggregate outside neurons (Taylor-Walker et al., 2016) (Figure 2E, F). This pattern of rapid cellular internalisation by small, soluble Aβ species such as oligomers was in keeping with the known biophysical characteristics of oligomeric Aβ (Williams et al., 2010; Benilova et al., 2012). Similarly Aβ assembly, in particular that of Aβ1–42, into larger structures was also consistent with the reported behavior of this aggregate-prone Aβ species (Jarrett et al., 1993; Ward et al., 2000; Lee et al., 2011). It is possible that numerous, small retinal Aβ deposits, which we detected in our mouse model were initially formed in this manner. On-going experiments in our laboratory are investigating the etiology of such retinal Aβ aggregates. Our work also revealed that internalized Aβ transiently impaired the microtubule-associated protein 2 (MAP-2) (Taylor-Walker et al., 2016), which normally maintains the spacing/gapping between adjacent microtubules (Teng et al., 2001; Harada et al., 2002). MAP-2 is also important for dendritic arbours (Vaillant et al., 2002) and contributes to long-term dendritic stability (Sudo and Baas, 2010). Chronic Aβ exposure could impair activities of neuroretinal cells during neuro-morphogenesis which occurs in post-mitotic neurons (Rich et al., 1997; Reye et al., 2002; Grandel et al., 2006; Pow and Sullivan, 2007). Indeed, MAP-2 is found co-localized to inner segments of abnormal photoreceptors with neurite sprouts, tortuous axons and abnormal nuclei in donor human AMD retinas (Pow and Sullivan, 2007). If chronic Aβ exposure induces MAP-2 impairment in retinal neurons, it could have implications for other neuronal activities including intracellular transport of cargos and synaptic plasticity.For instance, our earlier work revealed how cytoskeletal elements such as actin can modulate neurotransmission which underpins plasticity (Ratnayaka et al., 2011). We suggest that such subtle Aβ-driven mechanisms might be at play over many years to irreversibly compromise retinal neurotransmission.ese may not always be evident, but if retinal neurons are targeted by Aβ in this manner, it may explain why some AMD patients present primary photoreceptor pathology in the absence of any RPE damage (Bird et al., 2014).
Future Directions
Here we have reviewed the latest literature related to Aβ deposition in the senescent retina, and discussed potential mechanisms by which it could impair retinal health based on our fi ndings.e acute Aβ-injection mouse model we developed allows us to study dynamic aspects of Aβ-mediated pathophysiology in living retinas which cannot be studied using difficult to source fixed donor tissues. Thisin vivomodel also allows us to control the dosage and types of Aβ to which the retina is exposed, as well as to mimic the subtleties and specif i cities of the AMD phenotype to a remarkable extent.e results show striking similarities to early-intermediate AMD, reproducing features such as pigment abnormalities, disrupted, hyperplastic RPE, BrM breakage as well as loss of POS within a relatively short period. As such, our acute Aβ-injection mouse model represents a powerful tool in the arsenal to study AMD. Our discoveries into how Aβ impairs retinal neurons at single-cell resolution provide the fi rst insights into how the retina may become damaged over decade-long Aβ exposure. On-going experiments will delve further into functional consequences to reveal novel mechanisms which could form the basis for future treatments. Our experimental work and those of others thus reveal the extent to which Aβ in donor aged/AMD retinas could actually contribute to disease (Figure 3). The potential implications of removing retinal Aβ was demonstrated in an elegant study where ApoE4-HFC transgenic mice were rescued from visual defects by systemic Aβ-immunotherapy (Ding et al., 2008, 2011). The importance of Aβ in diagnosing AMD has also become evident, as plasma Aβ levels were recently demonstrated to be a promising biomarker for those suf f ering from late-stage AMD (Guymer et al., 2015). Taken together, these developments could reveal an as yet poorly def i ned but important disease mechanism in the retina, which could also be used to predict AMD and provide insights into its rate of progression.
Acknowledgments:We thank Mr. Thomas Freeman and Ms. Roshni Desai for their work on the Aβ-injection mouse model, Mr. George Taylor-Walker and Ms Jenny Scott for in vitro modelling experiments and Dr. David A. Johnston (Biomedical Imaging Unit, University of Southampton) for his expertise in imaging. We would also like to thank Dr. Helen S.K. Ratnayaka (Sussex Community NHS Foundation Trust) for reading the manuscript.
Author contributions:EK, RM, GG and HG collected the data. SAL, AJL and JAR drafted the manuscript.
Conficts of interest:Te authors report no conf l icts on interests.
Amoaku WM, Chakravarthy U, Gale R, Gavin M, Ghanchi F, Gibson J, Harding S, Johnston RL, Kelly S, Lotery A, Mahmood S, Menon G, Sivaprasad S, Talks J, Tufail A, Yang Y (2015) Def i ning response to anti-VEGF therapies in neovascular AMD. Eye (Lond) 29:721-731.
Anderson DH, Talaga KC, Rivest AJ, Barron E, Hageman GS, Johnson LV (2004) Characterization of beta amyloid assemblies in drusen: the deposits associated with aging and age-related macular degeneration. Exp Eye Res 78:243-256.
Anderson DH, Radeke MJ, Gallo NB, Chapin EA, Johnson PT, Curletti CR, Hancox LS, Hu J, Ebright JN, Malek G, Hauser MA, Rickman CB, Bok D, Hageman GS, Johnson LV (2010)e pivotal role of the complement system in aging and age-related macular degeneration: hypothesis re-visited. Prog Retin Eye Res 29:95-112.
Anderson PJ, Watts HR, Jen S, Gentleman SM, Moncaster JA, Walsh DT, Jen LS (2009) differential ef f ects of interleukin-1beta and S100B on amyloid precursor protein in rat retinal neurons. Clin Ophthalmol 3:235-242.
Atwood CS, Obrenovich ME, Liu T, Chan H, Perry G, Smith MA, Martins RN (2003) Amyloid-beta: a chameleon walking in two worlds: a review of the trophic and toxic properties of amyloid-beta. Brain Res Brain Res Rev 43:1-16.
Benilova I, Karran E, De SB (2012) The toxic Abeta oligomer and Alzheimer’s disease: an emperor in need of clothes. Nat Neurosci 15:349-357.
Bhutto I, Lutty G (2012) Understanding age-related macular degeneration (AMD): relationships between the photoreceptor/retinal pigment epithelium/Bruch’s membrane/choriocapillaris complex. Mol Aspects Med 33:295-317.
Bird AC, Phillips RL, Hageman GS (2014) Geographic atrophy: a histopathological assessment. JAMA Ophthalmol 132:338-345.
Bitan G, Fradinger EA, Spring SM, Teplow DB (2005) Neurotoxic protein oligomers--what you see is not always what you get. Amyloid 12:88-95.
Bradt BM, Kolb WP, Cooper NR (1998) Complement-dependent proinf l ammatory properties of the Alzheimer’s disease beta-peptide. J Exp Med 188:431-438.
Broersen K, Jonckheere W, Rozenski J, Vandersteen A, Pauwels K, Pastore A, Rousseau F, Schymkowitz J (2011) A standardized and biocompatible preparation of aggregate-free amyloid beta peptide for biophysical and biological studies of Alzheimer’s disease. Protein Eng Des Sel 24:743-750.
Cao L, Wang H, Wang F, Xu D, Liu F, Liu C (2013) Abeta-induced senescent retinal pigment epithelial cells create a proinf l ammatory microenvironment in AMD. Invest Ophthalmol Vis Sci 54:3738-3750.
Chew EY, Clemons TE, Agron E, Sperduto RD, Sangiovanni JP, Kurinij N, Davis MD (2013) Long-term effects of vitamins C and E, beta-carotene, and zinc on age-related macular degeneration: AREDS report no. 35. Ophthalmology 120:1604-1611.e1604.
Chiu CJ, Chang ML, Zhang FF, Li T, Gensler G, Schleicher M, Taylor A (2014) The relationship of major American dietary patterns to age-related macular degeneration. Am J Ophthalmol 158:118-127.
Cipriani V, Leung HT, Plagnol V, Bunce C, Khan JC, Shahid H, Moore AT, Harding SP, Bishop PN, Hayward C, Campbell S, Armbrecht AM, Dhillon B, Deary IJ, Campbell H, Dunlop M, Dominiczak AF, Mann SS, Jenkins SA, Webster AR, et al. (2012) Genome-wide association study of age-related macular degeneration identif i es associated variants in the TNXB-FKBPL-NOTCH4 region of chromosome 6p21.3. Hum Mol Genet 21:4138-4150.
Citron M, Westaway D, Xia W, Carlson G, Diehl T, Levesque G, Johnson-Wood K, Lee M, Seubert P, Davis A, Kholodenko D, Motter R, Sherrington R, Perry B, Yao H, Strome R, Lieberburg I, Rommens J, Kim S, Schenk D, et al. (1997) Mutant presenilins of Alzheimer’s disease increase production of 42-residue amyloid beta-protein in both transfected cells and transgenic mice. Nat Med 3:67-72.
Cunvong K, Huf f mire D, Ethell DW, Cameron DJ (2013) Amyloid-beta increases capillary bed density in the adult zebrafish retina. Invest Ophthalmol Vis Sci 54:1516-1521.
Dasari B, Prasanthi JR, Marwarha G, Singh BB, Ghribi O (2011) Cholesterol-enriched diet causes age-related macular degeneration-like pathology in rabbit retina. BMC Ophthalmol 11:22.
Davis MD, Gangnon RE, Lee LY, Hubbard LD, Klein BE, Klein R, Ferris FL, Bressler SB, Milton RC (2005)e Age-Related Eye Disease Study severity scale for age-related macular degeneration: AREDS Report No. 17. Arch Ophthalmol 123:1484-1498.
de Oliveira Dias JR, de Andrade GC, Kniggendorf VF, Novais EA, Maia A, Meyer C, Watanabe SE, Farah ME, Rodrigues EB (2016) Clinical and electrophysiological evaluation aer intravitreal ziv-af l ibercept for exudative age-related macular degeneration. Retina doi:10.1097/ IAE.0000000000001385.
DeMattos RB, Bales KR, Parsadanian M, O’Dell MA, Foss EM, Paul SM, Holtzman DM (2002) Plaque-associated disruption of CSF and plasma amyloid-beta (Abeta) equilibrium in a mouse model of Alzheimer’s disease. J Neurochem 81:229-236.
Dentchev T, Milam AH, Lee VM, Trojanowski JQ, Dunaief JL (2003) Amyloid-beta is found in drusen from some age-related macular degeneration retinas, but not in drusen from normal retinas. Mol Vis 9:184-190.
Ding JD, Lin J, Mace BE, Herrmann R, Sullivan P, Bowes RC (2008) Targeting age-related macular degeneration with Alzheimer’s disease based immunotherapies: anti-amyloid-beta antibody attenuates pathologies in an age-related macular degeneration mouse model. Vision Res 48:339-345.
Ding JD, Johnson LV, Herrmann R, Farsiu S, Smith SG, Groelle M, Mace BE, Sullivan P, Jamison JA, Kelly U, Harrabi O, Bollini SS, Dilley J, Kobayashi D, Kuang B, Li W, Pons J, Lin JC, Bowes RC (2011) Anti-amyloid therapy protects against retinal pigmented epithelium damage and vision loss in a model of age-related macular degeneration. Proc Natl Acad Sci U S A 108:E279-E287.
Duf f K, Eckman C, Zehr C, Yu X, Prada CM, Perez-tur J, Hutton M, Buee L, Harigaya Y, Yager D, Morgan D, Gordon MN, Holcomb L, Refolo L, Zenk B, Hardy J, Younkin S (1996) Increased amyloid-beta42(43) in brains of mice expressing mutant presenilin 1. Nature 383:710-713.
Dutescu RM, Li QX, Crowston J, Masters CL, Baird PN, Culvenor JG (2009) Amyloid precursor protein processing and retinal pathology in mouse models of Alzheimer’s disease. Graefes Arch Clin Exp Ophthalmol 247:1213-1221.
Elias FT, Silva EN, Belfort R, Jr., Silva MT, Atallah AN (2015) Treatment options for age-related macular degeneration: a budget impact analysis from the perspective of the Brazilian public health system. PLoS One 10:e0139556.
Ennis S, Jomary C, Mullins R, Cree A, Chen X, MacLeod A, Jones S, Collins A, Stone E, Lotery A (2008) Association between the SERPING1 gene and age-related macular degeneration: a two-stage case-control study. Lancet 372:1828-1834.
Ferris FL, Davis MD, Clemons TE, Lee LY, Chew EY, Lindblad AS, Milton RC, Bressler SB, Klein R (2005) A simplified severity scale for age-related macular degeneration: AREDS Report No. 18. Arch Ophthalmol 123:1570-1574.
Fisichella V, Giurdanella G, Platania CB, Romano GL, Leggio GM, Salomone S, Drago F, Caraci F, Bucolo C (2016) TGF-beta1 prevents rat retinal insult induced by amyloid-beta (1-42) oligomers. Eur J Pharmacol 787:72-77.
Friedman DS, O’Colmain BJ, Munoz B, Tomany SC, McCarty C, de Jong PT, Nemesure B, Mitchell P, Kempen J (2004) Prevalence of age-related macular degeneration in the United States. Arch Ophthalmol 122:564-572.
Fritsche LG, Chen W, Schu M, Yaspan BL, Yu Y,orleifsson G, Zack DJ, Arakawa S, Cipriani V, Ripke S, Igo RP Jr, Buitendijk GH, Sim X, Weeks DE, Guymer RH, Merriam JE, Francis PJ, Hannum G, Agarwal A, Armbrecht AM, et al. (2013) Seven new loci associated with age-related macular degeneration. Nat Genet 45:433-432.
Fritsche LG, Igl W, Bailey JN, Grassmann F, Sengupta S, Bragg-Gresham JL, Burdon KP, Hebbring SJ, Wen C, Gorski M, Kim IK, Cho D, Zack D, Souied E, Scholl HP, Bala E, Lee KE, Hunter DJ, Sardell RJ, Mitchell P, et al. (2015) A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat Genet 48:134-143.
Gibson J, Hakobyan S, Cree AJ, Collins A, Harris CL, Ennis S, Morgan BP, Lotery AJ (2012) Variation in complement component C1 inhibitor in age-related macular degeneration. Immunobiology 217:251-255.
Gordois A, Cutler H, Pezzullo L, Gordon K, Cruess A, Winyard S, Hamilton W, Chua K (2012) An estimation of the worldwide economic and health burden of visual impairment. Glob Public Health 7:465-481.
Goverdhan SV, Howell MW, Mullins RF, Osmond C, Hodgkins PR, Self J, Avery K, Lotery AJ (2005) Association of HLA class I and class II polymorphisms with age-related macular degeneration. Invest Ophthalmol Vis Sci 46:1726-1734.
Grandel H, Kaslin J, Ganz J, Wenzel I, Brand M (2006) Neural stem cells and neurogenesis in the adult zebraf i sh brain: origin, proliferation dynamics, migration and cell fate. Dev Biol 295:263-277.
Grunwald JE, Pistilli M, Ying GS, Maguire MG, Daniel E, Martin DF (2015) Growth of geographic atrophy in the comparison of age-related macular degeneration treatments trials. Ophthalmology 122:809-816.
Guo L, Salt TE, Luong V, Wood N, Cheung W, Maass A, Ferrari G, Russo-Marie F, Sillito AM, Cheetham ME, Moss SE, Fitzke FW, Cordeiro MF (2007) Targeting amyloid-beta in glaucoma treatment. Proc Natl Acad Sci U S A 104:13444-13449.
Guo LY, Alekseev O, Li Y, Song Y, Dunaief JL (2014) Iron increases APP translation and amyloid-beta production in the retina. Exp Eye Res 129:31-37.
Guymer R, Cipriani T, Rittenhouse KD, Lim L, Robman LD, Li W, Wang W, Deng S, Banerjee P (2015) Plasma levels of amyloid beta and other proinf l ammatory mediators in patients with age-related macular degeneration. Graefes Arch Clin Exp Ophthalmol 253:1347-1354.
Hahn P, Milam AH, Dunaief JL (2003) Maculas af f ected by age-related macular degeneration contain increased chelatable iron in the retinal pigment epithelium and Bruch’s membrane. Arch Ophthalmol 121:1099-1105.
Harada A, Teng J, Takei Y, Oguchi K, Hirokawa N (2002) MAP2 is required for dendrite elongation, PKA anchoring in dendrites, and proper PKA signal transduction. J Cell Biol 158:541-549.
He N, Jin WL, Lok KH, Wang Y, Yin M, Wang ZJ (2013) Amyloid-beta(1-42) oligomer accelerates senescence in adult hippocampal neural stem/progenitor cells via formylpeptide receptor 2. Cell Death Dis 4:e924.
Hebert LE, Scherr PA, Bienias JL, Bennett DA, Evans DA (2003) Alzheimer disease in the US population: prevalence estimates using the 2000 census. Arch Neurol 60:1119-1122.
Hoh KJ, Lenassi E, Jef f ery G (2010) Viewing ageing eyes: diverse sites of amyloid Beta accumulation in the ageing mouse retina and the up-regulation of macrophages. PLoS One 5:e13127.
Howlett DR, Bate ST, Collier S, Lawman A, Chapman T, Ashmeade T, Marshall I, Anderson PJ, Philpott KL, Richardson JC, Hille CJ (2011) Characterisation of amyloid-induced inf l ammatory responses in the rat retina. Exp Brain Res 214:185-197.
Isas JM, Luibl V, Johnson LV, Kayed R, Wetzel R, Glabe CG, Langen R, Chen J (2010) Soluble and mature amyloid fi brils in drusen deposits. Invest Ophthalmol Vis Sci 51:1304-1310.
Jarrett JT, Berger EP, Lansbury PT, Jr. (1993)e carboxy terminus of the beta amyloid protein is critical for the seeding of amyloid formation: implications for the pathogenesis of Alzheimer’s disease. Biochemistry 32:4693-4697.
Jef f eries WA, Price KA, Biron KE, Fenninger F, Pfeifer CG, Dickstein DL (2013) Adjusting the compass: new insights into the role of angiogenesis in Alzheimer’s disease. Alzheimers Reser 5:64.
Johnson LV, Leitner WP, Rivest AJ, Staples MK, Radeke MJ, Anderson DH (2002)e Alzheimer’s A beta -peptide is deposited at sites of complement activation in pathologic deposits associated with aging and age-related macular degeneration. Proc Natl Acad Sci U S A 99:11830-11835.
Khandhadia S, Cherry J, Lotery AJ (2012) Age-related macular degeneration. Adv Exp Med Biol 724:15-36.
Kim W, Hecht MH (2005) Sequence determinants of enhanced amyloidogenicity of Alzheimer A{beta}42 peptide relative to A{beta}40. J Biol Chem 280:35069-35076.
Kipfer-Kauer A, McKinnon SJ, Frueh BE, Goldblum D (2010) Distribution of amyloid precursor protein and amyloid-beta in ocular hypertensive C57BL/6 mouse eyes. Curr Eye Res 35:828-834.
Klein R, Klein BE, Tomany SC, Meuer SM, Huang GH (2002) Ten-year incidence and progression of age-related maculopathy:e Beaver Dam eye study. Ophthalmology 109:1767-1779.
Klein R, Cruickshanks KJ, Nash SD, Krantz EM, Nieto FJ, Huang GH, Pankow JS, Klein BE (2010)e prevalence of age-related macular degeneration and associated risk factors. Arch Ophthalmol 128:750-758.
Korb CA, Kottler UB, Wolfram C, Hoehn R, Schulz A, Zwiener I, Wild PS, Pfeif f er N, Mirshahi A (2014) Prevalence of age-related macular degeneration in a large European cohort: results from the population-based Gutenberg Health Study. Graefes Arch Clin Exp Ophthalmol 252:1403-1411.
Koronyo-Hamaoui M, Koronyo Y, Ljubimov AV, Miller CA, Ko MK, Black KL, Schwartz M, Farkas DL (2011) Identif i cation of amyloid plaques in retinas from Alzheimer’s patients and noninvasive in vivo optical imaging of retinal plaques in a mouse model. Neuroimage 54 Suppl 1:S204-217.
Krishnan T, Ravindran RD, Murthy GVS, Vashist P, Fitzpatrick KE,ulasiraj RD, John N, Maraini G, Camparini M, Chakravarthy U, Fletcher AE (2010) Prevalence of early and late age-related macular degeneration in India: The INDEYE Study. Invest Ophthalmol Vis Sci 51:701-707.
Kuperstein I, Broersen K, Benilova I, Rozenski J, Jonckheere W, Debulpaep M, Vandersteen A, Segers-Nolten I, Van Der Werf K, Subramaniam V, Braeken D, Callewaert G, Bartic C, D’Hooge R, Martins IC, Rousseau F, Schymkowitz J, De Strooper B (2010) Neurotoxicity of Alzheimer’s disease Abeta peptides is induced by small changes in the Abeta42 to Abeta40 ratio. EMBO J 29:3408-3420.
Lacor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, Wood M, Viola KL, Klein WL (2007) Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer’s disease. J Neurosci 27:796-807.
Lee J, Culyba EK, Powers ET, Kelly JW (2011) Amyloid-beta forms fibrils by nucleated conformational conversion of oligomers. Nat Chem Biol 7:602-609.
Lengyel I, Flinn JM, Peto T, Linkous DH, Cano K, Bird AC, Lanzirotti A, Frederickson CJ, van Kuijk FJ (2007) High concentration of zinc in sub-retinal pigment epithelial deposits. Exp Eye Res 84:772-780.
Liu C, Cao L, Yang S, Xu L, Liu P, Wang F, Xu D (2015) Subretinal injection of amyloid-beta peptide accelerates RPE cell senescence and retinal degeneration. Int J Mol Med 35:169-176.
Lois N, McBain V, Abdelkader E, Scott NW, Kumari R (2013) Retinal pigment epithelial atrophy in patients with exudative age-related macular degeneration undergoing anti-vascular endothelial growth factor therapy. Retina 33:13-22.
Lotery A, Trump D (2007) Progress in def i ning the molecular biology of age related macular degeneration. Hum Genet 122:219-236.
Luibl V, Isas JM, Kayed R, Glabe CG, Langen R, Chen J (2006) Drusen deposits associated with aging and age-related macular degeneration contain nonf i brillar amyloid oligomers. J Clin Invest 116:378-385.
Lynn SA, Goverdhan S, Munday R, Scott J, Freeman T, Johnston D, Lotery AJ, Ratnayaka JA (2016) A mouse model to study Aβ-driven pathology in the ageing retina. Invest Ophthalmol Vis Sci 57:6534-6534.
Malek G, Johnson LV, Mace BE, Saloupis P, Schmechel DE, Rickman DW, Toth CA, Sullivan PM, Bowes RC (2005) Apolipoprotein E allele-dependent pathogenesis: a model for age-related retinal degeneration. Proc Natl Acad Sci U S A 102:11900-11905.
Marmor MF, Wolfensberger T (1998)e Retinal Pigment Epithelium. Function and Disease. New York: Oxford.
Mc Donald JM, Savva GM, Brayne C, Welzel AT, Forster G, Shankar GM, Selkoe DJ, Ince PG, Walsh DM (2010)e presence of sodium dodecyl sulphate-stable Abeta dimers is strongly associated with Alzheimer-type dementia. Brain 133:1328-1341.
McKay GJ, Silvestri G, Chakravarthy U, Dasari S, Fritsche LG, Weber BH, Keilhauer CN, Klein ML, Francis PJ, Klaver CC, Vingerling JR, Ho L, De Jong PT, Dean M, Sawitzke J, Baird PN, Guymer RH, Stambolian D, Orlin A, Seddon JM, et al. (2011) Variations in apolipoprotein E frequency with age in a pooled analysis of a large group of older people. Am J Epidemiol 173:1357-1364.
McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, Bush AI, Masters CL (1999) Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Ann Neurol 46:860-866.
Minter MR, Taylor JM, Crack PJ (2016)e contribution of neuroinfl ammation to amyloid toxicity in Alzheimer’s disease. J Neurochem 136:457-474.
Mullins RF, Russell SR, Anderson DH, Hageman GS (2000) Drusen associated with aging and age-related macular degeneration contain proteins common to extracellular deposits associated with atherosclerosis, elastosis, amyloidosis, and dense deposit disease. FASEB J 14:835-846.
Noguchi A, Matsumura S, Dezawa M, Tada M, Yanazawa M, Ito A, Akioka M, Kikuchi S, Sato M, Ideno S, Noda M, Fukunari A, Muramatsu S, Itokazu Y, Sato K, Takahashi H, Teplow DB, Nabeshima Y, Kakita A, Imahori K, et al. (2009) Isolation and characterization of patient-derived, toxic, high mass amyloid beta-protein (Abeta) assembly from Alzheimer disease brains. J Biol Chem 284:32895-32905.
Ohno-Matsui K (2011) Parallel fi ndings in age-related macular degeneration and Alzheimer’s disease. Prog Retin Eye Res 30:217-238.
Owen CG, Fletcher AE, Donoghue M, Rudnicka AR (2003) How big is the burden of visual loss caused by age related macular degeneration in the United Kingdom? Br J Ophthalmol 87:312-317.
Owen CG, Jarrar Z, Wormald R, Cook DG, Fletcher AE, Rudnicka AR (2012)e estimated prevalence and incidence of late stage age related macular degeneration in the UK. Br J Ophthalmol 96:752-756.
Paraoan L, Ratnayaka A, Spiller DG, Hiscott P, White MR, Grierson I (2004) Unexpected intracellular localization of the AMD-associated cystatin C variant. Traf fi c 5:884-895.
Perez SE, Lumayag S, Kovacs B, Mufson EJ, Xu S (2009) Beta-amyloid deposition and functional impairment in the retina of the APPswe/ PS1DeltaE9 transgenic mouse model of Alzheimer’s disease. Invest Ophthalmol Vis Sci 50:793-800.
Perrin RJ, Fagan AM, Holtzman DM (2009) Multimodal techniques for diagnosis and prognosis of Alzheimer’s disease. Nature 461:916-922.
Ponte P, Gonzalez-DeWhitt P, Schilling J, Miller J, Hsu D, Greenberg B, Davis K, Wallace W, Lieberburg I, Fuller F (1988) A new A4 amyloid mRNA contains a domain homologous to serine proteinase inhibitors. Nature 331:525-527.
Pow DV, Sullivan RK (2007) Nuclear kinesis, neurite sprouting and abnormal axonal projections of cone photoreceptors in the aged and AMD-afflicted human retina. Exp Eye Res 84:850-857.
Ratnapriya R, Zhan X, Fariss RN, Branham KE, Zipprer D, Chakarova CF, Sergeev YV, Campos MM, Othman M, Friedman JS, Maminishkis A, Waseem NH, Brooks M, Rajasimha HK, Edwards AO, Lotery A, Klein BE, Truitt BJ, Li B, Schaumberg DA, et al. (2014) Rare and common variants in extracellular matrix gene Fibrillin 2 (FBN2) are associated with macular degeneration. Human Mol Genet 23:5827-5837.
Ratnayaka A, Marra V, Branco T, Staras K (2011) Extrasynaptic vesicle recycling in mature hippocampal neurons. Nat Commun 2:531.
Ratnayaka A, Paraoan L, Nelson G, Spiller DG, White MR, Hiscott P (2007a) Trafficking of osteonectin by retinal pigment epithelial cells: evidence for basolateral secretion. Int J Biochem Cell Biol 39:85-92.
Ratnayaka A, Paraoan L, Spiller DG, Hiscott P, Nelson G, White MR, Grierson I (2007b) A dual Golgi- and mitochondria-localised Ala-25Ser precursor cystatin C: an additional tool for characterising intracellular mis-localisation leading to increased AMD susceptibility. Exp Eye Res 84:1135-1139.
Ratnayaka JA, Lynn S (2016) Alzheimer’s-Related Amyloid Beta Peptide Aggregates in the Ageing Retina: Implications for Sight Loss and Dementia. Chapter 5:85-108. Croatia: Intech Publishing.
Ratnayaka JA, Serpell LC, Lotery AJ (2015) Dementia of the eye: the role of amyloid beta in retinal degeneration. Eye (Lond) 29:1013-1026.
Reye P, Sullivan R, Pow DV (2002) Distribution of two splice variants of the glutamate transporter GLT-1 in the developing rat retina. J Comp Neurol 447:323-330.
Rich KA, Zhan Y, Blanks JC (1997) Migration and synaptogenesis of cone photoreceptors in the developing mouse retina. J Comp Neurol 388:47-63.
Rudolf M, Curcio CA (2009) Esterified cholesterol is highly localized to Bruch’s membrane, as revealed by lipid histochemistry in wholemounts of human choroid. J Histochem Cytochem 57:731-739.
Rudolf M, Clark ME, Chimento MF, Li CM, Medeiros NE, Curcio CA (2008) Prevalence and morphology of druse types in the macula and periphery of eyes with age-related maculopathy. Invest Ophthalmol Vis Sci 49:1200-1209.
Sarks SH, Arnold JJ, Killingsworth MC, Sarks JP (1999) Early drusen formation in the normal and aging eye and their relation to age related maculopathy: a clinicopathological study. Br J Ophthalmol 83:358-368.
Selkoe DJ (2008) Soluble oligomers of the amyloid beta-protein impair synaptic plasticity and behavior. Behav Brain Res 192:106-113.
Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL (2007) Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci 27:2866-2875.
Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ (2008) Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med 14:837-842.
Sivaprasad S, Bird A, Nitiahpapand R, Nicholson L, Hykin P, Chatziralli I (2016) Perspectives on reticular pseudodrusen in age-related macular degeneration. Surv Ophthalmol 61:521-537.
Smith MA, Taneda S, Richey PL, Miyata S, Yan SD, Stern D, Sayre LM, Monnier VM, Perry G (1994) Advanced Maillard reaction end products are associated with Alzheimer disease pathology. Proc Natl Acad Sci U S A 91:5710-5714.
Sofat R, Casas JP, Webster AR, Bird AC, Mann SS, Yates JR, Moore AT, Sepp T, Cipriani V, Bunce C, Khan JC, Shahid H, Swaroop A, Abecasis G, Branham KE, Zareparsi S, Bergen AA, Klaver CC, Baas DC, Zhang K, et al. (2012) Complement factor H genetic variant and age-related macular degeneration: ef f ect size, modif i ers and relationship to disease subtype. Int J Epidemiol 41:250-262.
Soura V, Stewart-Parker M, Williams TL, Ratnayaka A, Atherton J, Gorringe K, Tuffin J, Darwent E, Rambaran R, Klein W, Lacor P, Staras K,orpe J, Serpell LC (2012) Visualization of co-localization in Abeta42-administered neuroblastoma cells reveals lysosome damage and autophagosome accumulation related to cell death. Biochem J 441:579-590.
Spaide RF (2009) Age-related choroidal atrophy. Am J Ophthalmol 147:801-810.
Spaide RF (2013) Outer retinal atrophy after regression of subretinal drusenoid deposits as a newly recognized form of late age-related macular degeneration. Retina 33:1800-1808.
Stone EM, Braun TA, Russell SR, Kuehn MH, Lotery AJ, Moore PA, Eastman CG, Casavant TL, Sheffield VC (2004) Missense variations in the fi bulin 5 gene and age-related macular degeneration. N Engl J Med 351:346-353.
Sudo H, Baas PW (2010) Acetylation of microtubules inf l uences their sensitivity to severing by katanin in neurons and fi broblasts. J Neurosci 30:7215-7226.
Tanzi RE, McClatchey AI, Lamperti ED, Villa-Komarof f L, Gusella JF, Neve RL (1988) Protease inhibitor domain encoded by an amyloid protein precursor mRNA associated with Alzheimer’s disease. Nature 331:528-530.
Taylor-Walker G, Lynn SA, Keeling E, Munday R, Johnston DA, Page A, Scott JA, Goverdhan S, Lotery AJ, Ratnayaka JA (2016)e Alzheimer’s-related amyloid beta peptide is internalised by R28 neuroretinal cells and disrupts the microtubule associated protein 2 (MAP-2). Exp Eye Res 153:110-121.
Teng J, Takei Y, Harada A, Nakata T, Chen J, Hirokawa N (2001) Synergistic ef f ects of MAP2 and MAP1B knockout in neuronal migration, dendritic outgrowth, and microtubule organization. J Cell Biol 155:65-76.
Townsend M, Shankar GM, Mehta T, Walsh DM, Selkoe DJ (2006) Effects of secreted oligomers of amyloid beta-protein on hippocampal synaptic plasticity: a potent role for trimers. J Physiol 572:477-492.
Tseng BP, Esler WP, Clish CB, Stimson ER, Ghilardi JR, Vinters HV, Mantyh PW, Lee JP, Maggio JE (1999) Deposition of monomeric, not oligomeric, Abeta mediates growth of Alzheimer’s disease amyloid plaques in human brain preparations. Biochemistry 38:10424-10431.
Vaillant AR, Zanassi P, Walsh GS, Aumont A, Alonso A, Miller FD (2002) Signaling mechanisms underlying reversible, activity-dependent dendrite formation. Neuron 34:985-998.
Walsh DT, Bresciani L, Saunders D, Manca MF, Jen A, Gentleman SM, Jen LS (2005) Amyloid beta peptide causes chronic glial cell activation and neuro-degeneration aer intravitreal injection. Neuropathol Appl Neurobiol 31:491-502.
Walsh DT, Montero RM, Bresciani LG, Jen AY, Leclercq PD, Saunders D, EL-Amir AN, Gbadamoshi L, Gentleman SM, Jen LS (2002) Amyloid-beta peptide is toxic to neurons in vivo via indirect mechanisms. Neurobiol Dis 10:20-27.
Wang J, Ohno-Matsui K, Morita I (2012a) Cholesterol enhances amyloid beta deposition in mouse retina by modulating the activities of Abeta-regulating enzymes in retinal pigment epithelial cells. Biochem Biophys Res Commun 424:704-709.
Wang J, Ohno-Matsui K, Morita I (2012b) Elevated amyloid beta production in senescent retinal pigment epithelium, a possible mechanism of subretinal deposition of amyloid beta in age-related macular degeneration. Biochem Biophys Res Commun 423:73-78.
Wang J, Zhu C, Xu Y, Liu B, Wang M, Wu K (2011) Development and expression of amyloid-beta peptide 42 in retinal ganglion cells in rats. Anat Rec (Hoboken ) 294:1401-1405.
Ward RV, Jennings KH, Jepras R, Neville W, Owen DE, Hawkins J, Christie G, Davis JB, George A, Karran EH, Howlett DR (2000) Fractionation and characterization of oligomeric, protofibrillar and fibrillar forms of beta-amyloid peptide. Biochem J 348 Pt 1:137-144.
Williams TL, Day IJ, Serpell LC (2010)e ef f ect of Alzheimer’s Abeta aggregation state on the permeation of biomimetic lipid vesicles. Langmuir 26:17260-17268.
Wong WL, Su X, Li X, Cheung CM, Klein R, Cheng CY, Wong TY (2014) Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: a systematic review and meta-analysis. Lancet Glob Health 2:e106-116.
Woodell A, Rohrer B (2014) A mechanistic review of cigarette smoke and age-related macular degeneration. Adv Exp Med Biol 801:301-307.
Ye H, Zhang Q, Liu X, Cai X, Yu W, Yu S, Wang T, Lu W, Li X, Jin H, Hu Y, Kang X, Zhao P (2014) Prevalence of age-related macular degeneration in an elderly urban chinese population in China: the Jiangning Eye Study. Invest Ophthalmol Vis Sci 55:6374-6380.
Yoshida T, Ohno-Matsui K, Ichinose S, Sato T, Iwata N, Saido TC, Hisatomi T, Mochizuki M, Morita I (2005)e potential role of amyloid beta in the pathogenesis of age-related macular degeneration. J Clin Invest 115:2793-2800.
Zhao T, Gao J, Van J, To E, Wang A, Cao S, Cui JZ, Guo JP, Lee M, Mc-Geer PL, Matsubara JA (2015) Age-related increases in amyloid beta and membrane attack complex: evidence of inf l ammasome activation in the rodent eye. J Neuroinf l ammation 12:121.
*< class="emphasis_italic">Correspondence to: J. Arjuna Ratnayaka, B.Sc., M.Phil., Ph.D., J.Ratnayaka@soton.ac.uk.
J. Arjuna Ratnayaka, B.Sc., M.Phil., Ph.D., J.Ratnayaka@soton.ac.uk.
orcid: 0000-0002-1027-6938 (J. Arjuna Ratnayaka)
10.4103/1673-5374.205083
Accepted: 2017-03-19
杂志排行

中国神经再生研究(英文版)的其它文章
- Recovery of multiply injured ascending reticular activating systems in a stroke patient
- Neuroprotective mechanism of Kai Xin San: upregulation of hippocampal insulin-degrading enzyme protein expression and acceleration of amyloid-beta degradation
- Mitomycin C induces apoptosis in human epidural scar fi broblasts after surgical decompression for spinal cord injury
- Exenatide promotes regeneration of injured rat sciatic nerve
- Recombinant human fi broblast growth factor-2 promotes nerve regeneration and functional recovery after mental nerve crush injury
- Ca2+involvement in activation of extracellular-signalregulated-kinase 1/2 and m-calpain after axotomy of the sciatic nerve