盖塔病毒分子进化及其迁徙
2017-04-26李元元李晓龙付士红高晓艳雷雯雯王环宇王桂琴梁国栋
李元元,刘 红,李晓龙,付士红,高晓艳,雷雯雯,吕 志,何 英,王环宇,王桂琴,梁国栋
盖塔病毒分子进化及其迁徙
李元元1,2,刘 红3,李晓龙2,付士红2,高晓艳2,雷雯雯2,吕 志2,何 英2,王环宇2,王桂琴1,梁国栋2
目的 了解世界各地分离盖塔病毒分子进化特征及其时空迁徙。方法 用Clustal X1.83、MegaAlign和Gene DOC软件对测定的核苷酸和推测的氨基酸序列进行比较分析,用MEGA 6.0软件绘制系统发生树,采用BEAST v 1.8.1软件包里的Bayesian Stochastic Search Variable Selection(BSSVS)程序进行盖塔病毒的空间动力学分析。结果 盖塔病毒E2基因全长均为1 266 nt,编码422 aa,其核苷酸和氨基酸同源性分别为94.5%~100%和96.4%~100%。病毒分子进化分析发现,盖塔病毒不存在蚊虫,马匹和猪等宿主动物和媒介的种属和地域分布差异。生物信息学分析结果显示,盖塔病毒起源于马来西亚,此后依次传播至日本、中国、韩国以及蒙古国和俄罗斯等地。结论 自1955年首次分离盖塔病毒至2014年间分离的盖塔病毒E2基因序列较为稳定,未发现病毒基因组之间存在种属及地域分布差异,并且该病毒已经从热带地区传播到欧亚大陆多个国家和地区,因此加强盖塔病毒及人畜动物感染的检测和监测至关重要。
盖塔病毒;分子进化;时空迁徙
盖塔病毒(Getah virus, GETV)于1955 年首次在马来西亚捕获的库蚊标本分离到,命名为Getah virus 其原株型为MM2021[1]。此后研究显示,盖塔病毒(Getah virus)属于披膜病毒科(Togaviridae)甲病毒属(Alphavirus)病毒,为蚊虫传播虫媒病毒[2]。盖塔病毒可以引起家畜疾病,如马的发热、皮疹、后腿水肿及淋巴结肿大,并能引起猪的致死性疾病和生殖障碍等[3-4],因此盖塔病毒被确定为重要动物源性病原体[5-6]。此外,在健康人血清样本和发热患者血清标本中均能检测到盖塔病毒中和抗体[7-10],表明该病毒也可以引起人类感染。
盖塔病毒自1955年在马来西亚分离之后,在日本、东南亚和澳大利亚等地,从三带喙库蚊、刺忧伊蚊等多种蚊虫标本和生病的马匹和猪的血液标本分离到盖塔病毒[3-4,11]。目前,已经约13个国家或地区发现盖塔病毒的存在,地域分布遍及大洋洲的澳大利亚[12]、马来西亚[1]、日本[13]及欧亚大陆的中国[7]、蒙古国[14]和俄罗斯[14]等。我国于1964年首次在海南分离到盖塔病毒(M1株)[7],并且进入21世纪以来,相继在中国多个省市采集的蚊虫标本中分离到多株盖塔病毒[15]。截止到目前(2016年11月),GenBank共收录近50株盖塔病毒的基本信息,包括日本分离株10株,韩国7株,马来西亚、蒙古、俄罗斯和中国台湾分离株各1株,其余分离株均来自中国大陆地区,因此盖塔病毒已成为我国以至于亚洲大陆地区的“新发虫媒病毒”。因此了解该病毒的分子进化特征及其时空动力迁徙,对于该病毒的预防和控制十分重要。本研究对序列信息最全的自1955—2014年世界各地分离的盖塔病毒E2基因序列进行分子遗传和生物信息学分析,以期了解病毒的分子进化特征及时空动态,为预防盖塔病毒的传播提供重要依据。
1 材料和方法
1.1 盖塔病毒E2基因序列数据集的建立 从GenBank数据库中下载所有盖塔病毒E2基因序列(截止到2016年11月),病毒基因序列包括1955—2014年世界各地不同国家、不同吸血昆虫和不同宿主动物标本中分离的26株盖塔病毒(表1)。包括我国6个省市或地区不同蚊种中分离的11株病毒,马来西亚首次分离的病毒株(MM2021),日本在不同宿主中分离的9株病毒,以及4株韩国猪标本中分离的病毒和俄罗斯、蒙古蚊虫分离的盖塔病毒株。病毒株在地域上跨越了北纬3°—60°,东经38°—140°的范围。
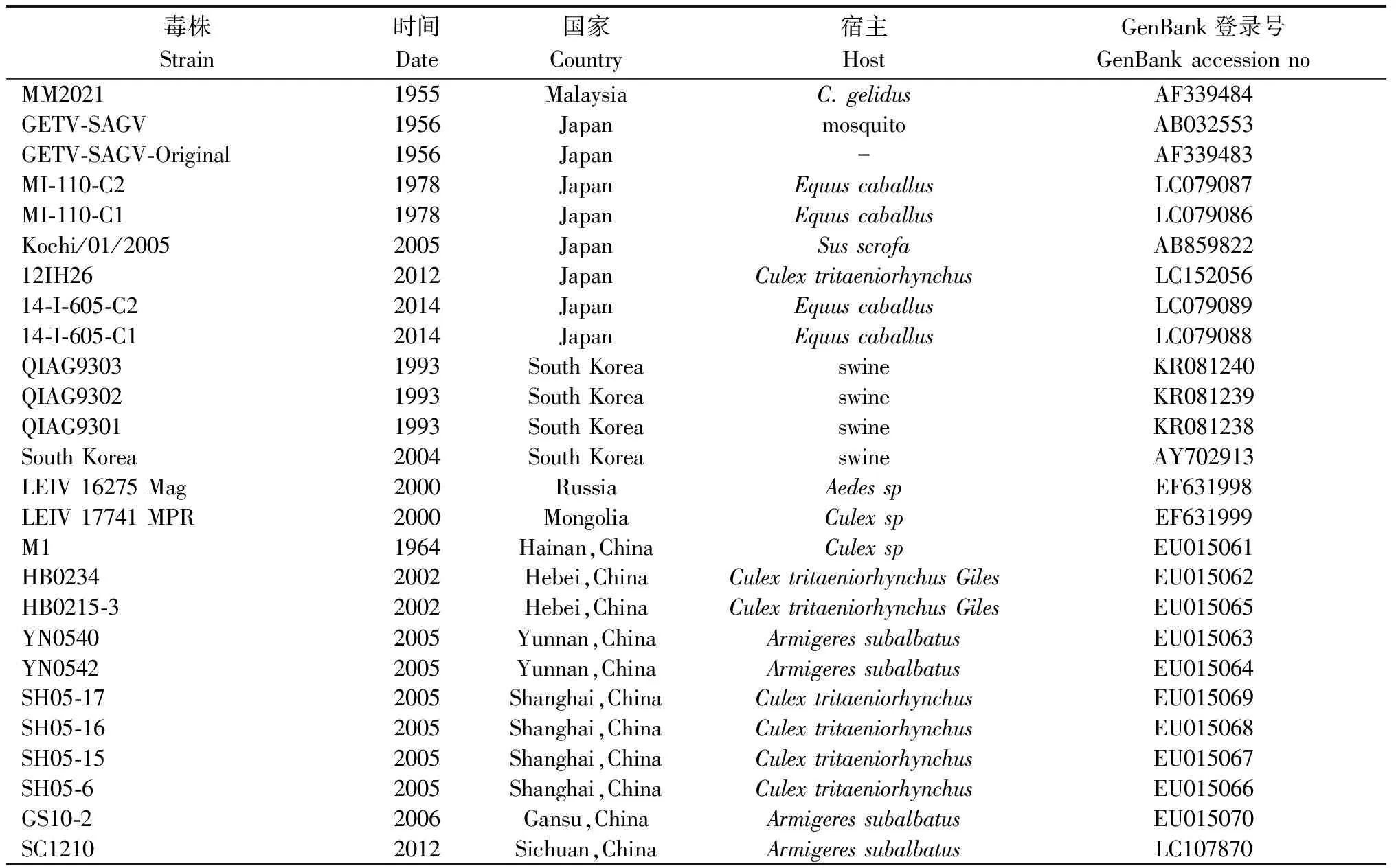
表1 本研究所使用的盖塔病毒基因组序列信息
Note:The sign “-” represents no detail information
1.2 盖塔病毒E2基因序列分析 将GETV 数据集的序列,使用ClustalX 1.8软件进行病毒核苷酸序列比对;使用BioEdit(version 7.0.5.3)和DNASTAR(5.00)软件包中的MegAlign 软件进行核苷酸和氨基酸序列的比对和同源性分析;使用Gene DOC软件进行核苷酸和氨基酸差异度分析;使用MEGA 6.0软件完成基于Neighbour-joining(NJ)方法的进化树绘制,Bootstrap值设定为1 000[16]。
1.3 盖塔病毒的空间动力学分析 采用BEAST v 1.8.1软件包里的Bayesian Stochastic Search Variable Selection (BSSVS)程序进行盖塔病毒的空间动力学研究。采用SPREAD软件对BEAST软件生成的地理迁徙分析结果进行可视化,最后用Google Earth v.6.2.2软件展示盖塔病毒的地理迁徙分析结果[17-18]。
2 结 果
2.1 盖塔病毒基因序列分析 对1955—2014年不同地域、不同宿主分离的26株盖塔病毒E2基因核苷酸和氨基酸序列分析,结果表明(表2):26株盖塔病毒核苷酸和氨基酸的同源性分别在94.5%~100%和96.4%~100%之间,提示盖塔病毒E2基因序列较为保守。氨基酸位点分析表明(表3),盖塔病毒E2基因全长均为1 266 nt,编码422 aa,不同地域、不同宿主来源的病毒编码区长度不存在差异。与1955年马来西亚原始株(MM2021)相比,其他25盖塔病毒分离株均存在7个共同的氨基酸差异位点,分别位于E27(丝氨酸S→苯丙氨酸F)、E90(苏氨酸T→缬氨酸V)、E102(丙氨酸A→缬氨酸V)、E122(异亮氨酸I→苏氨酸T)、E213(精氨酸R→丝氨酸S)、E314(丙氨酸A→缬氨酸V)、E323(谷氨酸E→天冬氨酸D,甘肃分离株GS10-2除外)。此外,与马来西亚原始分离株(MM2021)相比其他盖塔病毒分离株还存在一些氨基酸差异位点,这些差异位点分散在E2基因不同位置(表3)。

表2 盖塔病毒E2基因核苷酸和氨基酸同源性比较(%)
注:右上部代表盖塔病毒E2基因核苷酸序列的同源性,左下部分代表盖塔病毒E2氨基酸序列的同源性
Note:The upper right part represents the homologous rate of nucleotide sequence of viral E2 gene. The lower left part represents the homologous rate of amino acid sequence of viral E2 gene
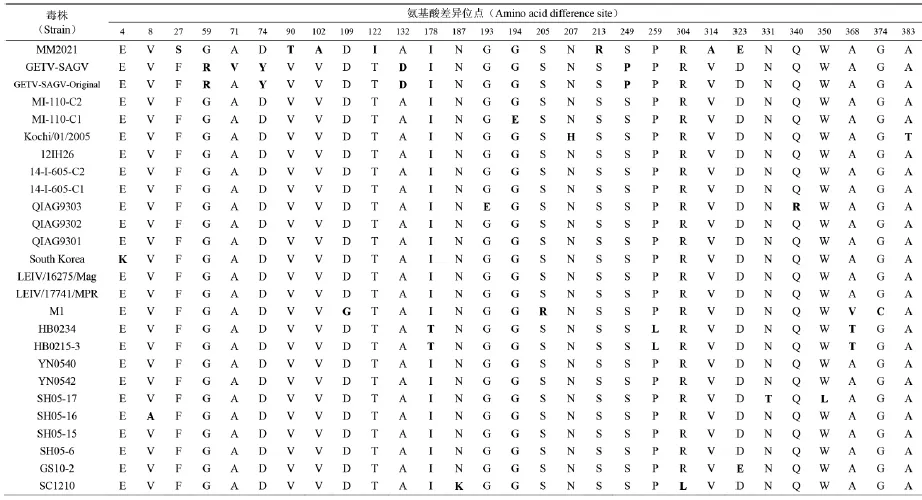
表3 盖塔病毒E2基因氨基酸差异位点分析
2.2 盖塔病毒系统进化分析 将国内外分离的所有盖塔病毒的E2基因序列构建基于NJ方法的系统进化树(图1)。结果显示,位于盖塔病毒进化树根部的种群为1955年马来西亚蚊虫中分离的盖塔病毒,紧接着是日本分离的SAGV种群和2000年俄罗斯蚊虫中分离的盖塔病毒,两者独立成簇,处于不同的进化分枝。距离盖塔病毒进化分枝根部较远的病毒种群,包括1964-2014年不同宿主,不同地域分离的盖塔病毒,该病毒种群位于同一进化簇中,是毒株数量最多的病毒进化分枝。这一分枝包括中国,韩国,蒙古和日本大部分地区分离的病毒,病毒分离地域跨度大,从北纬19°至48°,是最年轻的病毒种群也是传播距离最远的病毒种群。

图1 盖塔病毒E2基因序列的系统进化分析Fig.1 Phylogenetic analysis of the E2 gene sequences of GETV
2.3 盖塔病毒的空间动力学分析 本研究采用Spread分析方法进行盖塔病毒时空动力学分析,结果显示(图2),马来西亚、日本和中国云南省是3个主要的盖塔病毒播散来源。盖塔病毒存在如下可能的播散路线(图2):1)1917年,从马来西亚播散到日本;2)1960年,从日本播散到中国海南省;3)1975年,从日本播散到中国云南省;4)1990年代,从日本播散到蒙古及韩国;以云南省为播散源,播散至俄罗斯地区,并在中国广泛散播开来。
3 讨 论
自1955年分离到第1株盖塔病毒至2014年已经分离到数十株盖塔病毒,时间跨度超过60年,分离毒株地域分布涉及北纬3°至北纬60°地区,东经38°至东经140°地区,可见盖塔病毒在亚欧大陆及太
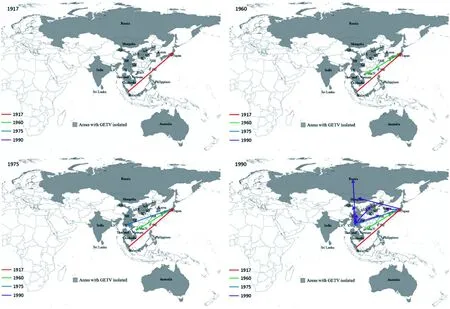
图2 盖塔病毒的传播路线Fig.2 Route of transmission of the GETV
平洋岛屿具有非常广泛的分布。盖塔病毒已多次在马和猪的标本中分离[3-4,13],提示盖塔病毒在自然界,特别是在家养动物中具有宿主。众所周知,蚊虫是盖塔病毒的传播媒介,自首次在马来西亚采集的雪背库蚊分离到盖塔病毒后,已经在4属9种蚊虫标本中分离到,分别为雪背库蚊[1]、三带喙库蚊[15,19-23]、伪杂鳞库蚊[24]、棕头库蚊[20]、环带库蚊[20]、中华按蚊[20]、白纹伊蚊[11]、刺扰伊蚊(GenBank登录号:JN410944、JN410945)和阿蚊[15,23]。以上结果提示,库蚊可能是盖塔病毒主要传播媒介。
目前已经有13个国家从多种蚊虫和马匹以及猪标本分离到约50株盖塔病毒,并且已经发现盖塔病毒可以引起马匹和猪的疾病[3-4]。1970年代以来在马来西亚、北澳大利亚、亚洲和太平洋岛国地区的健康人血清和发热患者血清标本中检测到盖塔病毒中和抗体阳性[8-10]。1992年中国海南省盖塔病毒血清流行病学研究结果显示,当地128份发热患者血清标本盖塔病毒中和抗体阳性率为10.9%(14/128)[7]。以上结果提示在马来西亚、北澳大利亚,亚洲、太平洋岛国和中国海南岛地区存在盖塔病毒在人畜动物中的感染。
鉴于盖塔病毒E2基因编码病毒外膜蛋白,是病毒与宿主细胞受体与配体相互作用部位[25],因此使用E2基因序列做病毒分子进化分析最能够代表盖塔病毒与吸血昆虫和宿主动物之间的分子进化趋势。盖塔病毒E2基因核苷酸与氨基酸同源性分析结果显示,自1955年至2014年世界各地,从不同蚊种,不同动物(马匹,猪)分离盖塔病毒的同源性在94.5%~100%和96.4%~100%,提示盖塔病毒外膜蛋白基因较为保守。但是病毒E2基因氨基酸差异位点分析发现,与1955年马来西亚蚊虫分离的盖塔病毒原型病毒株(MM2021)相比,其他25盖塔病毒分离株均存在7个共同的氨基酸差异位点(表3)。由于除MM2021病毒分离株以外,其他病毒均分离自马来西亚以外的地区或国家,中国、蒙古、俄罗斯、日本等地,而这些地区属于欧亚大陆地区,属于温带气候,因此,这7个共同突变的氨基酸是否是盖塔病毒从热带地区传播到中南亚和欧亚大陆后病毒基因组发生变异的分子基础,还需要开展进一步研究。
系统进化分析显示,日本1956年分离两株SAGE病毒处于1955年马来西亚原始盖塔病毒分离株与1964年以后近代分离盖塔病毒之间,提示SAGE病毒可能属于最初盖塔病毒向近代分离株进化过程中的过渡病毒毒株(图1)。此外,无论从蚊虫和马匹或者猪标本分离的盖塔病毒均位于系统进化树不同位置,未出现不同媒介或宿主的专一性分布,也未见到地域上的专一性分布。
此前对乙脑病毒迁徙时间研究结果显示[26],乙脑病毒从东南亚(马来西亚和菲律宾)起源后,逐渐向北形成4条传播路线。南部路线(泰国和我国云南省),东部离线(经我国东南沿海向东北部传到日本),中部路线(经云南省传向我国中部地区,如贵州、湖南、湖北)和西部路线(云南省传至我国四川、西藏及印度地区等)。本研究发现盖塔病毒的研究结果与此前对乙脑病毒研究结果有相似之处。本研究结果显示马来西亚、日本、和我国云南省是盖塔病毒迁徙事件发起的重要源泉地区(图2)。马来西亚发生的迁徙事件可以到达日本,提示马来西亚是盖塔病毒向东亚传播的源泉地区,这与马来西亚所处的地域气候因素有重要的关系。马来西亚属于热带地区,年平均气温高,降雨量大,适宜多种蚊虫的栖息和繁殖等。盖塔病毒在这一地区经过当地的鸟类、野生动物或当地饲养的家猪和马(猪和马是盖塔病毒的宿主[3-4,13])大量繁殖,扩增,然后通过候鸟形成该传播路线。
日本发生的迁徙事件可以使盖塔病毒到达中国海南省,云南省,韩国及蒙古国,提示日本在维持盖塔病毒在东部沿海和中国内陆省份及韩国、蒙古国循环的过程中起到了重要的作用。推测盖塔病毒可能借助东风通过蚊虫传播到内陆或沿海地区。
云南省作为盖塔病毒迁徙事件发起的重要地区,主要向俄罗斯、河北、上海、甘肃、四川等省份传播,提示云南省是维持盖塔病毒在中国内陆省份的循环源泉地区。我国已经在云南省的蚊虫调查中多次分离到盖塔病毒[11,15,20-21,24],提示云南省蚊虫中具有较高盖塔病毒携带率。云南省位于我国西南部,系北回归线以南,亚洲大陆向东南亚半岛过渡地带,本地区热量丰富,终年温暖,四季常青。云南蚊虫种类多,蚊虫密度高,居民普遍养猪,而且以散养为主,为盖塔病毒提供了充分的扩散宿主。也为盖塔病毒通过蚊虫传播到其它省份提供了条件。
空间动力学分析结果显示盖塔病毒的播散存在频繁的迁徙事件,而且既包括近距离的邻省间的迁徙事件,也包括远距离的跨国界间的迁徙事件。但是本研究盖塔病毒基因序列相对较少,因此该地理传播路线仅代表盖塔病毒的播散趋势,并非盖塔病毒的确切播散路线。
我国已经在10多个省市的多种蚊虫标本分离到盖塔病毒,但是一直没有发现人或动物感染盖塔病毒疾病流行的报道,也未在人畜动物标本中分离到盖塔病毒。因此要加强盖塔病毒在人畜动物中,特别是对马匹和家猪盖塔病毒感染的检测和监测,以便及时发现盖塔病毒感染在人畜动物中的流行,避免该疾病流行所引起的各种经济损失。
[1] Karabatsos N. International catalogue of arthropod-borne viruses[M]. 3rd ed. San Antonio:American Society for Tropical Medicine and Hygiene. 1985.
[2] Weaver SC, Frey TK, Huang HV, et al. Virus taxonomy. Eighth report of the international committee on taxonomy of viruses[M]. London:Elsevier/Academic Press, 2005:999-1008.
[3] Manabu N, Hiroshi B, Koji T, et al. Getah virus infection among Racehorses, Japan, 2014[J]. Emerg Infect Dis, 2015, 21(5):883-885.
[4] Yago K, Hagiwara S, Kawamura H, et al. A fatal case in Newborn piglets with Getah virus infection:isolation of the virus[J]. Jpn J Vet Sci, 1987, 49(6):989-994.
[5] KumanoInido T, Wada R, Kanenlanl T, et al. Clinical and virological observatiom on swine experimentally infected with Getah virus[J]. Vet Microbiol, 1988, 16(3):295-301.
[6] Kumanomido T, Kamada M, Wada R, et al. Pathogenicity for horses of original sagiyama virus, a member of the Getah vires group[J]. Vet Microbiol, 1988, 17(4):367-373.
[7] Li XD, Qiu FX, Yang H, et al. Isolation of Getah virus from mosquitos collected on Hainan Island, China, and results of a serosurvey[J]. Southeast Asian J Trop Med Public Health, 1992, 23(4):730-734.
[8] Marchette NJ, Rudnick A, Garcia R. Alphaviruses in Peninsular Malaysia:II. Serological evidence of human infection[J]. Southeast Asian J Trop Med Public Health, 1980, 11(1):14-23.
[9] Kanamitsu M, Taniguchi K, Urasawa S, et al. Geographic distribution of arbovirus antibodies in indigenous human populations in the Indo-Australian archipelago[J]. Am J Trop Med Hyg, 1979, 28(2):351-363.
[10] Tesh RB, Gajdusek DC, Garruto RM, et al. The distribution and prevalence of group A arbovirus neutralizing antibodies among human populations in Southeast Asia and the Pacific islands[J]. Am J Trop Med Hyg, 1975, 24(4):664-675.
[11] Zhou XF, Feng ZL, Hu TS, et al. Isolation and identification of Getah virus from Aedes albopictus in Yunnan[J]. Chin J Health Lab Technol, 2012, 22(1):103-106. (in Chinese)
周晓芳,冯子良,胡挺松,等.云南白纹伊蚊盖塔病毒的分离鉴定[J].中国卫生检验杂志,2012, 22(1):103-106.
[12] Fukunaga Y, Kumanomido T, Kamada M. Getah virus as an equine pathogen[J]. Vet Clin North Am Equine Pract, 2000, 16(3):605-617.
[13] Wekesa SN, Inoshima Y, Murakami K, et al. Genomic analysis of some Japanese isolates of Getah virus[J]. Vet Microbiol, 2001, 83(2):137-146.
[14] L’vov SD, Gromashevski’i VL, Aristova VA, et al. Isolation of Getah virus (Togaviridae,Alfavirus) strains in North-Eastern Asia[J]. Vopr Virusol, 2000, 45(5):14-18. (in Russian).
[15] Zhai YG, Wang HY, Sun XH, et al. Complete sequence characterization of isolates of Getah virus (genus Alphavirus, family Togaviridae) from China[J]. J General Virol, 2008, 89(6):1446-1456.
[16] Lei WW, Guo XF, Fu SH, et al. Isolation of Tibet orbivirus, TIBOV, from culicoides collected in Yunnan, China[J]. PLoS One, 2015, 10(8):e0136257. DOI:10.1371/journal.pone.0136257
[17] Lemey P, Rambaut A, Drummond AJ, et al. Bayesian phylogeography finds its roots[J]. PLoS Comput Biol, 2009, 5(9):e1000520. DOI:10.1371/journal.pcbi.1000520
[18] Bielejec F, Rambaut A, Suchard MA, et al. SPREAD:spatial phylogenetic reconstruction of evolutionary dynamics[J]. Bioinformatics, 2011, 27(20):2910-2912.
[19] Wang HQ, Liu WB, Yang DR, et al. Isolation and identification of arboviruses in Hebei Province[J]. Chin J Exp Clin Virol, 2006, 20(1):52-55. (in Chinese)
王焕琴,刘卫滨,杨冬荣,等.河北省虫媒病毒分离鉴定[J].中华实验和临床病毒学杂志,2006, 20(1):52-55.
[20] Feng Y, Fu SH, Yang WH, et al. Molecular characterization of mosquito-borne viruses strains newly isolated in Western Yunnan Province along the border of China and Myanmar,China[J]. Intl J Virol, 2014, 21(5):193-198. (in Chinese)
冯云,付士红,杨卫红,等.云南省西部边境新分离蚊媒病毒分子生物学特征分析[J].国际病毒学杂志,2014, 21(5):193-198.
[21] Feng Y, Zhang HL, Fu SH, et al. Investigation on mosquitoes and mosquito-borne viruses in Dehong prefecture, Yunnan province, 2007 and 2010[J]. Chin J Epidemiol, 2014, 35(5):528-532. (in Chinese)
冯云,张海林,付士红,等.云南省德宏州2007 年和2010 年蚊虫及蚊媒病毒调查[J].中华流行病学杂志,2014, 35(5):528-532.
[22] Chen WX, Wang HY, Fu SH, et al. Partial genome molecular characteristics of Getah virus newly isolated in China[J]. Chin J Microbiol Immunol, 2010, 30(5):399-404. (in Chinese)
陈维欣,王环宇,付士红,等.我国新分离盖塔病毒的部分基因组分子特征研究[J].中华微生物学和免疫学杂志,2010, 30(5):399-404.
[23] Li MH, Fu SH, Feng Y, et al. Investigation of arboviruses in different regions of Guizhou province,China in 2008[J]. Chin J Vector Biol Control, 2012, 23(5):417-420. (in Chinese)
李铭华,付士红,冯云,等.贵州省不同地区2008年虫媒病毒调查[J].中国媒介生物学及控制杂志,2012, 23(5):417-420.
[24] Feng Y, Li SG, Zhang HL, et al. Molecular characterization of Japanese encephalitis virus and Getah virus strains newly isolated in Tengchong County,Yunnan Province,China[J]. Chin J Zoonoses, 2014, 30(4):353-363. DOI:10.3969/cjz.j.issn.100-694.014.04.003(in Chinese)
冯云,李胜国,张海林,等.云南腾冲县新分离乙型脑炎和盖塔病毒的分子生物学特征[J].中国人兽共患病学报,2014, 30(4):353-363.
[25] Jin Q. Medical molecular virology[M]. Beijing:Science Press, Medical Publishing Center. 2001:401-429. (in Chinese)
金奇.医学分子病毒学[M].北京:科学出版社, 2001:401-429.
[26] Gao XY, Liu H, Wang HY, et al. Southernmost Asia is the source of Japanese encephalitis virus (genotype 1) diversity from which the viruses disperse and evolve throughout Asia[J]. PLoS Negl Trop Dis, 2013, 7(9):e2459. DOI:10.1371/journal.pntd.0002459
s:Liang Guo-dong, Email:gdliang@hotmail.com
Phylogenctics of Getah virus and its migration
LI Yuan-yuan1,2, LIU Hong3, LI Xiao-long2, FU Shi-hong2, GAO Xiao-yan2, LEI Wen-wen2,LYU Zhi2, HE Ying2, WANG Huan-yu2, WANG Gui-qin1, LIANG Guo-dong2
(1.ShanxiMedicalUniversity,Taiyuan030001,China; 2.StateKeyLaboratoryforInfectiousDiseasePreventionandControl,NationalInstituteforViralDiseaseControlandPrevention,ChineseCenterforDiseaseControlandPrevention,Beijing100052,China; 3.ShandongUniversityofTechnology,Zibo255000,China)
In order to investigate the molecular evolution and spatio-temporal migration of Getah viruses (GETV) isolated around the world, the nucleotide and deduced amino acid sequence of GETVs were analyzed and phylogenetic trees were constructed by using informatics software including ClustalX1.83, MegaAlign, GeneDOC and Mega6.0. The Bayesian Stochastic Search Variable Selection (BSSVS) program in the BEAST v 1.8.1 software package was used to analyze the spatial dynamics of the Getah virus. Results showed that the full-length of Getah virus E2 gene consists of 1 266 nueleotides, encoding 422 amino acids. And the homology of nucleotide and amino acid were 94.5%—100% and 96.4%—100% respectively. The molecular evolution analysis revealed that there were no species and geographical distribution difference existing among GETV host animals (e.g. horses and pigs) and vectors (e.g. mosquitoes). Bioinformatics analysis showed that GETV originated in Malaysia, then it was spread to Japan, China, South Korea, Mongolia, Russia, etc. GETV E2 gene was relatively stable since GETV was first isolated in 1955. The differences of species and geographical distribution did not exist among GETV host animals and vectors, and the virus has spread from tropical regions to Eurasian continent. Thus, strengthening the detection and monitoring of GETV and its infections in humans and livestock is critical.
Getah virus; molecular evolution; spatial and temporal migration
10.3969/j.issn.1002-2694.2017.04.001
国家自然科学基金重大项目(No.81290342)资助。李元元和刘红同等贡献
梁国栋,Email:gdliang@hotmail.com
1.山西医科大学,太原 030001; 2.中国疾病预防控制中心病毒病预防控制所/传染病预防控制国家重点实验室,北京 100052; 3.山东理工大学,淄博 255000
Supported by the National Natural Science Foundation of China (No. 81290342).Li Yuan-yuan and Liu Hong contributed equally
R373
A
1002-2694(2017)04-0293-07
2016-12-21 编辑:梁小洁
