超声波促进吡咯并哒嗪类化合物的合成
2017-04-25丁俊熊陈石泉王世范
丁俊熊,陈石泉,王世范
(海南大学 海洋学院,海南 海口 570228)
超声波促进吡咯并哒嗪类化合物的合成
丁俊熊,陈石泉,王世范
(海南大学 海洋学院,海南 海口 570228)
以β酮酸酯为起始原料,经亚硝化、Knorr缩合和硝酸铈铵氧化得到了5个吡咯醛.在40Hz,200W超声波作用下,5个吡咯醛与水合肼反应生成5个吡咯并哒嗪化合物.结果显示:全部化合物通过熔点,经TLC,IR和1H-NMR表征,结果和目标化合物相同.在超声波辅助作用下,利用吡咯醛和水合肼反应制备吡咯并哒嗪化合物,具有操作简单、能耗低、合成效率高等优点.其中2个吡咯醛(2d,2e)和4个吡咯并哒嗪化合物(3a,3c-3e)为首次报道.
吡咯; 吡咯并哒嗪; 超声波; 合成
哒嗪环是二氮六元杂环,广泛应用于医药、农药、材料领域[1-5],是很多药物的重要组成部分[6-7].据报道,吡咯并哒嗪结构的化合物中存在着多样的生物活性物质,如抗幽门螺杆菌[8]、抗肿瘤[9]、抗病毒[10]、抗激酶[11]等的活性物质.吡咯并哒嗪类化合物具有独特的结构特征和广泛的生物活性,化学合成此类化合物有实际的意义.已有报道,在超声波辅助下能提高化学合成反应的速率[12],相比于传统的加热回流方法,该法有操作简单,耗能低,合成效率高等优点[13].
本研究旨在探讨于超声波促进下合成新的吡咯并哒嗪类化合物,即以β酮酸酯为起始原料,经硝化反应后,在超声波辅助下利用Knorr缩合得到取代的吡咯1a-1e,然后经过硝酸铈铵氧化得到吡咯醛2a-2e,再在超声波辅助下与水合肼反应得到吡咯并哒嗪类化合物3a-3e.
目标化合物1a-1e, 2a-2e和3a-3e的合成反应路线见图1:

图1 化合物1a-1e, 2a-2e和3a-3e的合成路线
目标化合物1a-1e, 2a-2e和3a-3e的取代基结构见表1.
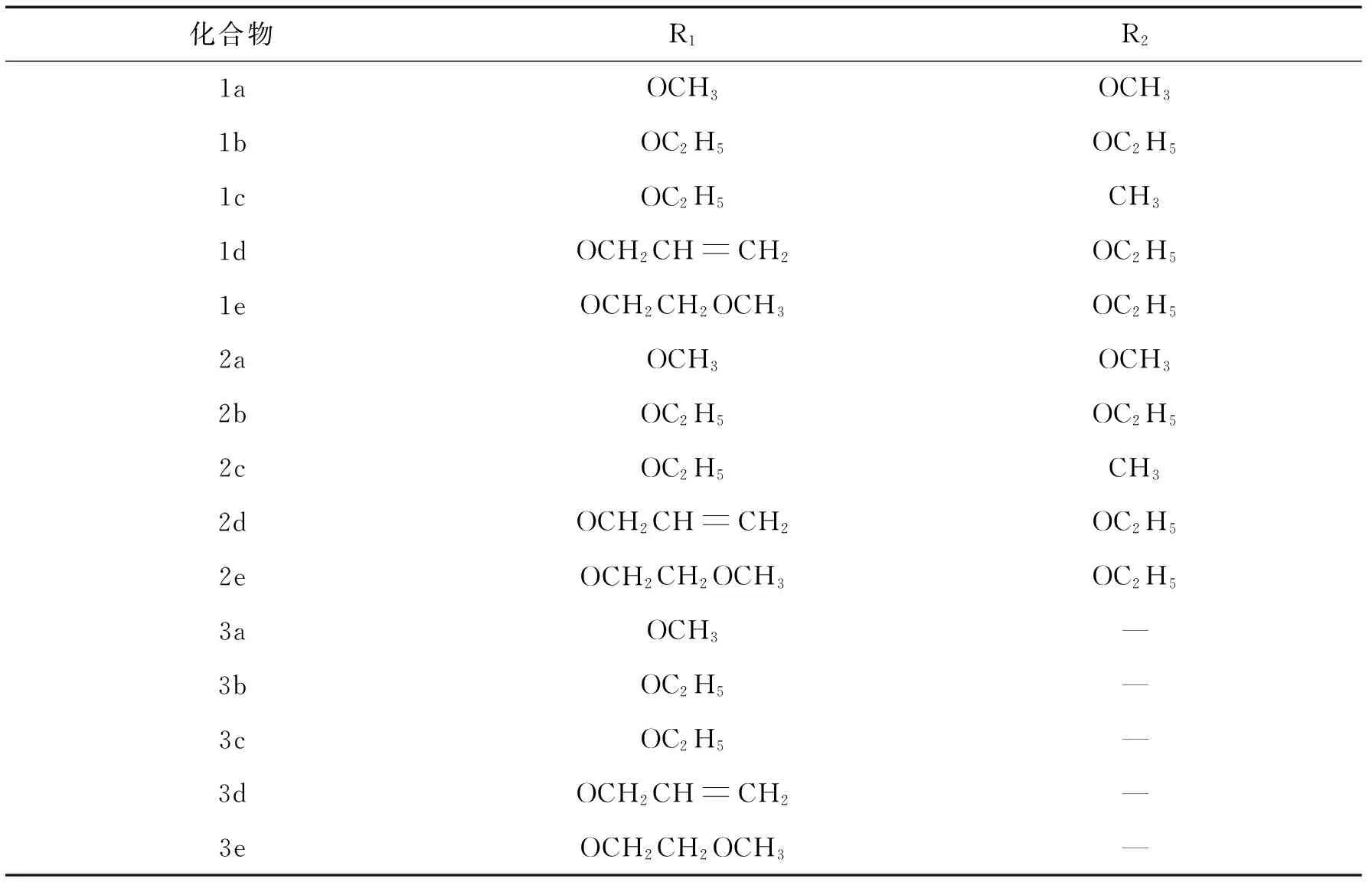
表1 目标化合物1a-1e,2a-2e,3a-3e
1 实验部分
1.1 仪器与试剂 熔点测定采用X4数字显微熔点仪(温度未校正);1H-NMR采用Bruker400/500MHz超导核磁共振仪(瑞士布鲁克公司)测定;IR采用ThermoiS10(美国赛默飞世尔科技公司).试剂均为市售分析纯,根据需要进行纯化.反应进程使用TCL薄板跟踪.
1.2 合成实验
1.2.1 化合物1a-1e的合成 参考文献[14]进行改进并合成.称取NaNO21.9g,将其配成饱和水溶液,逐滴加入到盛有25mmol(2.9g)乙酰乙酸甲酯和50mL冰醋酸的反应瓶中.滴加时在冰水浴中搅拌,并注意滴速要慢,以溶液颜色无明显变化为宜,滴毕,转至室温中搅拌2h, 可得硝化产物.然后加入25mmol(2.9g)乙酰乙酸甲酯,于超声波辅助下反应(40kHz, 500W),缓慢且少量多次地加完6g锌粉,65 ℃反应0.5h.终止反应,反应液加到冰水中,搅拌后使固体充分析出.抽滤、用水洗去水溶性杂质(至pH≈7)、干燥并结晶得无色针状晶体3,5-二甲基-2,4-二甲氧羰基-1H-吡咯.
以相似的方法合成化合物1b-1e
1a:(3,5-二甲基-2,4-二甲氧羰基-1H-吡咯).收率83%.m.p.167~168℃;1HNMR(CDCl3)δ:2.51(s,3H,吡咯上CH3),2.55(s,3H,吡咯上CH3),3.82 (s,3H,OCH3),3.86(s,3H,OCH3),9.10(s,1H,NH);IR(KBr)ν:3 297,2 951,2 851,1 705,1 677,1 566,1 515,1 282cm-1.
1b:(3,5-二甲基-2,4-二乙氧羰基-1H-吡咯).收率83%.m.p.133~135℃;1HNMR(CDCl3)δ:1.36 (t,J=7.1Hz,6H,2CH2CH3),2.51(s,3H,吡咯上CH3),2.56(s,3H,CH3),4.31 (q,J=7.1Hz,4H,2CH2),8.80 (s,1H,NH);IR(KBr)ν:3 267,2 984,2 933,1 689,1 669,1 572,1 483,1 263cm-1.
1c:(3,5-二甲基-4-乙酰基-2-乙氧羰基-1H-吡咯).收率65%.m.p.130~132℃;1HNMR(CDCl3):δ:1.37 (t,J=7.1Hz,3H,CH2CH3),2.45(s,3H,吡咯上CH3),2.52(s,3H,吡咯上CH3),2.59(s,3H,COCH3),4.33(q,J=7.1Hz,2H,CH3CH2),9.11(s,1H,NH);IR(KBr)ν:3 284,2 986,2 934,1 651,1 556,1 515,1 280cm-1.
1d:(3,5-二甲基-4-乙氧羰基-2-烯丙氧羰基-1H-吡咯).收率77%.m.p.110~112℃;1HNMR(d6-DMSO)δ:1.26(t,J=7.1Hz,3H,CH2CH3),2.41(s,3H,吡咯上CH3),2.47(s,3H,吡咯上CH3), 4.17(q,J=7.1Hz,2H,CH2CH3),4.74(dt,J1=5.4Hz,J2=1.4Hz,2H,CH2CHCH2O),5.25(dq,J1=10.5Hz,J2=1.4Hz,1H,CH2CH),5.37(dq,J1=17.2Hz,J2=1.6Hz,1H,CH2CH),5.96-6.06 (m,1H,CH2CH),11.90(s,1H,NH);IR(KBr)ν:3 269,2 977,2 932,1 697,1 673,1 572,1 487,1 275cm-1.
1e:(3,5-二甲基-4-乙氧羰基-2-甲氧乙氧羰基-1H-吡咯).收率74%.m.p.103~106℃;1HNMR(CDCl3)δ:1.36(t,J=7.1Hz,3H,CH2CH3),2.51(s,3H,吡咯上CH3),2.58(s,3H,吡咯上CH3),3.42(s,3H,OCH3),3.69(t,J=4.6Hz,2H,CH3OCH2CH2O),4.29(q,J=7.1Hz,2H,CH2CH3),4.42(t,J=4.6Hz,2H,CH3OCH2CH2O),8.92(s,1H,NH);IR(KBr)ν:3 304,2 981,2 931,1 708,1 673,1 570,1 438,1 281cm-1.
1.2.2 化合物2a-2e的合成 参考文献[15]进行改进合成.在锥形瓶中加入60mL四氢呋喃,溶解15mmol化合物1,依次倒入60mL冰醋酸和60mL蒸馏水.称取硝酸铈铵60mmol(33.0g),分多次慢慢加入至溶液中,在冰水浴中搅拌至全溶.此时颜色较深,于室温再搅拌约2~3h,待颜色淡去且反应完成后,加到水中,搅拌后放置一段时间,挥发四氢呋喃,使固体充分析出.抽滤、多次水洗至pH≈7、干燥并结晶,得无色针状晶体3-甲基-5-甲酰基吡咯羧酸酯.
2a:(3-甲基-5-甲酰基-2,4-二甲氧羰基-1H-吡咯).收率72%.m.p.137~138℃;1HNMR(CDCl3)δ:2.60(s,3H,吡咯上CH3),3.93(s,6H,2OCH3),9.87(s,1H,NH),10.25(s,1H,CHO);IR(KBr)ν:3 279,2 962,2 902,1 708,1 673,1 548,1 496,1 250cm-1.
2b:(3-甲基-5-甲酰基-2,4-二乙氧羰基-1H-吡咯).收率73%.m.p.119~121℃;1HNMR(CDCl3)δ:1.40(t,J=7.1Hz,6H,2CH2CH3),2.61(s,3H,吡咯上CH3),4.39(q,J=7.1Hz,4H,2OCH2),9.87(s,1H,NH),10.27(s,1H,CHO);IR(KBr)ν:3 275,2 982,2 935,1 716,1 680,1 552,1 463,1 268cm-1.
2c: (3-甲基-5-甲酰基-4-乙酰基-2-乙氧羰基-1H-吡咯).收率60%.m.p.105~107℃;1HNMR(CDCl3)δ:1.40(t,J=7.1Hz,3H,CH2CH3),2.60(s,3H,吡咯上CH3), 2.63(s,3H,COCH3),4.39(q,J=7.1Hz,2H,CH3CH2),9.92(s,1H,NH),10.13(s,1H,CHO);IR(KBr)ν:3 272,2 994,2 939,1 684,1 661,1 539,1 457,1 266cm-1.
2d: (3-甲基-5-甲酰基-4-乙氧羰基-2-烯丙氧羰基-1H-吡咯).收率62%.m.p.105~107 ℃;1HNMR(CDCl3)δ:1.41(t,J=7.1Hz,3H,CH2CH3),2.62(s,3H,吡咯上CH3),4.40(q,J=7.1Hz,2H,CH3CH2),4.82(dt,J1=5.4Hz,J2=1.4Hz,2H,CH2=CHCH2O),5.33(dq,J1=10.5Hz,J2=1.4Hz,1H,CH2=CH),5.41(dq,J1=17.0Hz,J2=1.4Hz,1H,CH2=CH),5.96-6.06(m,1H,CH2=CH),9.90 (s,1H,NH),10.27(s,1H,CHO);IR(KBr)ν:3 275,2 985,2 938,1 712,1 698,1 650,1 552,1 259cm-1.
2e: (3-甲基-5-甲酰基-4-乙氧羰基-2-甲氧乙氧羰基-1H-吡咯).收率60%.m.p.105~107 ℃;1HNMR(CDCl3)δ:1.41(t,J=7.1Hz,3H,CH2CH3),2.62(s,3H,吡咯上CH3),3.42(s,3H,OCH3),3.71(t,J=4.6Hz,2H,CH3OCH2CH2O),4.40(q,J=7.1Hz,2H,CH2CH3),4.47(t,J=4.6Hz,2H,CH3OCH2CH2O),10.01(s,1H,NH),10.27(s,1H,CHO);IR(KBr)ν:3 266,2 989,2 934,1 712,1 673,1 555,1 488,1 274cm-1.
1.2.3 化合物3a-3e的合成 反应瓶中加入1mmol3-甲基-5-甲酰基吡咯羧酸酯,冰醋酸6mL左右.取水合肼(80%)1~4mL(依反应情况增减),在反应过程中分多次加完(加样注意安全).反应液呈浅黄色,超声波辅助(40kHz, 200W)反应,加热至70 ℃左右,反应液渐渐变成白色浊液,TLC跟踪进程,反应约0.5h.所有的物质加到大量冰水中,静置,使固体物质充分析出,抽滤、洗涤除去水溶性杂质,干燥,得粉末粗产物.粗产物用乙醇洗涤,再用丙酮洗涤,干燥得白色粉末吡咯并哒嗪化合物.
3a:(3-甲基-4-羰基-2-甲氧羰基-1H-吡咯[2,3-d]并哒嗪).收率81%.m.p.327~328 ℃;1HNMR(d6-DMSO)δ:2.62(s,3H,吡咯上CH3),3.87(s,3H,OCH3),8.04(s,1H,哒嗪环上CH),12.26(s,1H, 哒嗪环上NH),12.54(s,1H,吡咯环上NH);IR(KBr)ν:3 259,3 181,3 113,2 963,1 691,1 649cm-1.
3b:(3-甲基-4-羰基-2-乙氧羰基-1H-吡咯[2,3-d]并哒嗪).收率82%.m.p.313~314 ℃;1HNMR(d6-DMSO)δ:1.34 (t,J=7.1Hz,3H,CH2CH3),2.62(s,3H,吡咯上CH3),4.34(q,2H,J=7.1Hz,CH2CH3),8.05(s,1H,哒嗪环上CH),12.28(s,1H,哒嗪环上NH),12.50(s,1H,吡咯环上NH);IR(KBr)ν:3 102,2 975,2 909,1 703,1 657cm-1.
3c:(3,4-二甲基-2-乙氧羰基-1H-吡咯[2,3-d]并哒嗪).收率77%.m.p.280~281℃;1HNMR(d6-DMSO)δ:1.36(t,J=7.1Hz,3H,CH2CH3),2.73(s,3H,吡咯上CH3),2.89(s,3H,哒嗪环上CH3),4.38(q,J=7.1Hz,2H,CH2CH3),9.15(s,1H,哒嗪环上CH),12.47(s,1H,吡咯环上NH);IR(KBr)ν:2 987,2 843,2 600,1 818,1 704cm-1.
3d:(3-甲基-4-羰基-2-烯丙氧羰基-1H-吡咯[2,3-d]并哒嗪).收率75%.m.p.288~290 ℃;1HNMR(d6-DMSO)δ:2.63(s,3H,吡咯上CH3),4.84(dt,J1=5.4Hz,J2=1.4Hz,2H,CH2CHCH2O),5.30(dq,J1=10.5Hz,J2=1.4Hz,1H,CH2=CH),5.43(dq,J1=17.0Hz,J2=1.4Hz,1H,CH2CH),6.00-6.10(m,1H,CH2CH),8.06(s,1H,哒嗪环上CH),12.31(s,1H,哒嗪环上NH),12.57(s,1H,吡咯环上NH);IR(KBr)ν:3 299,2 928,2 818,1 689,1 640cm-1.
3e: (3-甲基-4-羰基-2-(甲氧乙基)羰基-1H-吡咯[2,3-d]并哒嗪).收率76%.m.p.271~272 ℃;1HNMR(d6-DMSO)δ:2.62(s,3H,吡咯上CH3),3.30(s,3H,OCH3),3.66(t,J=4.4Hz,2H,CH3OCH2CH2O),4.42(t,J=4.4Hz,2H,CH3OCH2CH2O),8.06(s,1H,哒嗪环上CH),12.30(s,1H,哒嗪环上NH),12.53(s,1H,吡咯环上NH);IR(KBr)ν:3 283,2 880,2 821,1 684,1 624cm-1.
2 结果与讨论
2.1 1a-1e的合成 本文采用Knorr合成法合成吡咯酯类化合物,其反应过程分为两步,第一步是β酮酸酯硝化反应,第二步是硝化后产物与β酮酸酯进行环化反应生成吡咯环.亚硝化反应需在低温下进行,因温度过高容易发生副反应,因此,本文采用冰浴,将反应温度控制在0~5 ℃.然而,环化反应要求较高的温度,文献[14]是在冰醋酸中加热回流反应.本文中la-le的合成是在超声波辅助下于冰醋酸体系中合成,反应在65 ℃左右即可顺利进行,这可使反应更加快速、安全、高效.
第一步的硝化反应要完全,如果反应不完全,则第二步副反应很多.第一步的硝化反应约1~3h可反应完全;第二步环化反应需控制反应时间,要避免反应时间过短而导致关环不完全,也要避免反应时间过长而引起副产物的发生.反应过程用TLC跟踪,第二步环化反应大约需要0.5h可反应完全.
2.2 2a-2e的合成 该步反应是将吡咯酯α位的甲基氧化成醛基,属于氧化反应,本文选取硝酸铈铵作为氧化剂.硝酸铈铵Ce(NH4)2(NO3)6具有选择性较好,反应温和等优点.在较低温度时,可将甲基氧化成醛基,在较高温度时,生成的醛基会进一步氧化生成羧酸.硝酸铈铵在酸性介质中氧化性更强,其氧化反应一般遵循自由基反应机理,反应过程中需要水的参与.经过实验的探索与改进,本文采用了体积比为4∶ 4∶ 4的THF-HAc-H2O溶液作为反应体系.
由于该反应属于放热反应,氧化过程伴随着大量热量的释放,不仅存在着安全隐患,而且反应温度过高会使副反应增多,因此,该反应需在较低温度下进行.本文采用冰浴和室温相结合的方法,初始反应体系置于冰浴中,分多次慢慢加入硝酸铈铵,待其完全溶解后,记录颜色并撤去冰浴,再改为室温下搅拌,这样可以避免氧化过度,且可加快反应速度,同时也增加了安全性.在氧化过程中,初始反应体系的颜色较深,于室温中搅拌反应1.5~2.5h,其颜色变淡,表明氧化反应基本完成(根据颜色的变化和TLC跟踪来判断反应完成情况).
2.3 3a-3e的合成 该步反应首先进行亲核加成,然后再缩合环化.水合肼一端的NH2基进攻吡咯醛5位上的醛羰基并缩合,另一端的NH2基进攻吡咯醛4位上的酯羰基并环化,形成吡咯并哒嗪化合物.水合肼具有碱性、吸湿性和毒性,在湿空气中会形成白雾,因此在加水合肼时应插入反应液中滴加,这样可减少与空气的接触,从而减少损失,减小对人体的危害.在超声波辅助反应过程中由于水合肼损耗较多,因此需加入过量的水合肼,经试验,水合肼的用量在1~4mL时(TLC跟踪进程,依据反应情况适量增减水合肼的用量),其产率较高.水合肼有一定的危险性,因此,在实验过程及操作中务必注意安全.
传统方法采用加热回流[16]来合成吡咯[2,3-d]并哒嗪化合物,反应需要在较高温度下进行.如果反应温度不够,则反应较慢且哒嗪关环不完全;如果反应温度过高,则水合肼的体系的危险性增大.因此,本文采用超声波辅助法合成吡咯[2,3-d]并哒嗪化合物,反应温度比传统的加热回流反应温度低,反应速度较快,耗能较低,反应更为高效.经试验,在超声波促进下反应温度在70 ℃左右时,哒嗪关环反应快、产率高.
3 结 论
以β酮酸酯为起始原料,通过亚消化反应、Knorr缩合反应和氧化反应可获得吡咯醛,然后在超声波促进下,用吡咯醛与水合肼反应可得到5个吡咯并哒嗪化合物(3a-3e).实验表明,超声波促进合成吡咯并哒嗪类化合物是一种操作简单、耗能低、反应高效的合成方法.
[1]HuFZ,ZhangGF,LiuB,etal.Anewmethodforthesynthesisandherbicidalactivityof3-phenoxy-6-( 1H-(substituted)pyrazol-1-yl)pyridazines[J].JournalofHeterocyclicChemistry,2009,46(46):584-590.
[2]DeebA,MouradE,ElenanyD.PyridazineDerivativesandRelatedCompounds,Part22:Synthesis,Reactions,andInsecticidalActivityof3-Amino5, 6-diaryl-1H-pyrazolo[3,4-c]pyridazines[J].PhosphorusSulfur&Silicon&theRelatedElements,2009,185(1):222-231.
[3]SiddiquiAA,MishraR,ShaharyarM,etal.Triazoleincorporatedpyridazinonesasanewclassofantihypertensiveagents:Design,synthesisandinvivoscreening[J].Bioorganic&MedicinalChemistryLetters,2011,42(28):1023-1026.
[4] 胡有洪,楼丽广,林世军,等.一类以N桥键的哒嗪酮类化合物在制备抗肿瘤的药物中的用途[P].CN102133217A,2011.
[5]MiaoS,AppletonA,BergerN,etal. 6,13-Diethynyl-5,7,12,14-tetraazapentacene[J].Chemistry—AEuropeanJournal,2009,15(20):4990-4993.
[6] 陆坤宏. 哒嗪酮合成工艺的研究[D].南京:南京工业大学,2003.
[7] 王腾,王礼琛,董颖.哒嗪酮类化合物的药用研究近况[J].药学进展,2006,30(6):246-251.
[8]HaruoI,MasahikoH,NobuhikoS.Pharmaceuticalscontainingpyrrolo-pyridazinederivativesandtheiruseasantiulceragents[P].JP2003119140,2003.
[9]MalinkaW,RedzickaA,LozachO.NewDerivativesofPyrrolo[3,4-d]pyridazinoneandTheirAnticancerEffects[J].ILFarmaco,2004,59(6):457-462.
[10]SunZX;LiLS;WSE.Preparationofpyrro[1,2-b]pyridazinonederivativesusefulintreatinginfectionsbyhepatitisCvirus[P].WO2007150001,2007.
[11] 邓炳初, 冯君, 张蕾. 吡咯并哒嗪类衍生物及其制备方法和用途[P].WO2007082470,2007.
[12]AshokkumarM.Thecharacterizationofacousticcavitationbubbles-anoverview[J].UltrasonSonochem,2011,18(4):864-872.
[13]LévêqueJM,CravottoG.Microwaves,powerultrasound,andionicliquids,anewsynergyingreenorganicsynthesis[J].ChimiaInternationalJournalforChem,2006,60(6):313-320.
[14] 朱琰婷,单衍强, 阎琨,等. 超声波促进合成新型吡咯α,β-不饱和酮[J].合成化学,2015,23(10):963-966.
[15]BellIM,StirdivantSM,AhernJ,etal.Biochemicalandstructuralcharacterizationofanovelclassofinhibitorsofthetype1insulin-likegrowthfactorandinsulinreceptorkinases[J].Biochemistry,2005,44(27):9430-9440.
[16]ChenSQ,JiangK,WangSF.Ethyl-3-methyl-4-oxo-4,5-dihydro-1H-pyrrolo[2,3-d]pyridazine-2-carboxylate[J].ActaCryst.,2010,66:o259.
Synthesis of Pyrrole[2,3-d]Pyridazine Derivatives Under Ultrasound Irradiation
Ding Junxiong, Chen Shiquan, Wang Shifan
(College of Ocean, Hainan University, Haikou 570228, China)
In the report, taken β keto acid esters as the starting material, five compounds of pyrrole aldehydes were synthetized by nitrosation, Knorr condensation and oxidation with ammonium ceric nitrate. Under ultrasound irradiation(40Hz, 200W), five pyrrole[2,3-d]pyridazine compounds were synthesized by the reaction of pyrrole aldehydes and hydrazine hydrates.All the compounds were characterized by m.p.,TLC,IR and1H-NMR.Compared with the conventional synthesis of pyrrole[2,3-d]pyridazine compounds, the current progress indicates that the preparation of pyrrole[2,3-d]pyridazine compounds through pyrrole aldehydes and hydrazine hydrate under ultrasound irradiation, has advantages of simple operation, low energy consumption and high efficiency of synthesis. After verification, two compounds of pyrrole aldehydes (2d-2e) and four compounds of pyrrole alcohols (3a,3c-3e) are new compounds.
pyrrole; Pyrrole pyridazine; Ultrasound; synthesis
2016-11-04
国家自然科学基金项目 (21162006);海南省中西部高校提升综合实力工作基金项目资助
丁俊熊(1990-),男,浙江武义人,硕士研究生,研究方向为药物化合物,E-mail:Ding_junxiong@163.com
王世范(1963-),男,安徽和县人,教授,博士生导师, E-mail:wangsf777@163.com
1004-1729(2017)01-0031-06
O
ADOl:10.15886/j.cnki.hdxbzkb.2017.0007
