反相高效液相色谱法测定八珍颗粒中阿魏酸含量
2017-04-10刘海燕
刘海燕
(河南省周口市食品药品检验所,河南 周口 466000)
·检验检测·
反相高效液相色谱法测定八珍颗粒中阿魏酸含量
刘海燕
(河南省周口市食品药品检验所,河南 周口 466000)
目的建立测定八珍颗粒中阿魏酸含量的反相高效液相色谱(RP-HPLC)法。方法色谱柱采用AgilentHc-C18柱(250mm×4.6mm,5 m),流动相为0.085%磷酸溶液-乙腈(83∶17),流速为1.0mL/min,检测波长为316 nm,柱温为35℃。结果阿魏酸进样量在0.069 7~0.836 4 g范围内与峰面积线性关系良好(r=0.999 85),平均回收率为97.97%,RSD为1.39%(n=6)。结论该方法可行、准确、重复性好,可作为八珍颗粒中阿魏酸的质量控制方法。
反相高效液相色谱法;八珍颗粒;阿魏酸;含量测定
八珍颗粒收载于2015年版《中国药典(一部)》[1]464,由熟地黄、当归、党参、炒白术、炒白芍、茯苓、川芎、炙甘草等中药组方,具有补气益血功效,用于治疗气血两虚、面色萎黄、食欲不振、四肢乏力、月经过多等症[1-2]。其中当归为主药,具有补血活血、调经止痛的功效[1]133,其主要成分有挥发油类、香豆素类、有机酸类、糖类、黄酮类、氨基酸、维生素和其他微量元素[3-5],具有很高的药用价值。有机酸类中主要成分阿魏酸为当归的指标成分[6]。原标准中只对芍药苷的含量作为控制指标,为了更好地控制八珍颗粒的质量,参考文献[7-10],采用高效液相色谱法测定产品中阿魏酸的含量,经方法学验证,该方法操作简便,专属性强,重复性好,阴性对照无干扰。现报道如下。
1 仪器与试药
1.1 仪器
LC-2010C型全自动高效液相色谱仪(日本岛津公司);CPA-225D型电子分析天平(赛多利斯科学仪器<北京>有限公司);明澈T-D24UV电水系统(法国西格玛公司);KQ-500型超声波震荡器(江苏省昆山市超声仪器有限公司。
1.2 试药
阿魏酸对照品(中国食品药品检定研究院,批号为0773-9910);八珍颗粒样品(四川禾润制药有限公司,批号为151202,160210,规格为每袋3.5 g)。乙腈和甲醇为色谱纯,磷酸为分析纯,水超纯化水。
2 方法与结果
2.1 色谱条件
色谱柱:AgilentHc-C18柱(250mm×4.6mm,5μm),柱温:35℃;流动相:0.085%磷酸溶液-乙腈(83∶17);流速:1.0m L/min;进样量:10μL;检测波长:316 nm。
2.2 溶液制备
对照品溶液:精密称取阿魏酸对照品6.97 mg,置50 m L棕色容量瓶,加70%甲醇超声溶解并稀释至刻度,摇匀,作为对照品贮备液;精密量取5m L,置10 mL容量瓶,加70%甲醇至刻度,摇匀,即得。
供试品溶液:取装量差异项下的本品内容物,研细,取1.049 5g,置具塞锥形瓶中,精密加入70%甲醇20m L,密塞,称定质量,加热回流30min,放冷,再称定质量,用70%甲醇补足减失的质量,摇匀,静置,取上清液,滤过,取续滤液,作为供试品溶液。
阴性对照品溶液:按处方比例及制备工艺制备不含当归的阴性样品,按供试品溶液制备方法制备阴性对照品溶液。
2.3 方法学考察
专属性试验:取上述3种溶液各10μL,进样测定,记录色谱图。对照品溶液色谱图中阿魏酸峰的理论板数为9 161,供试品溶液色谱图中,阿魏酸与邻分峰达到基线分离,峰形对称,分离效果良好,且阴性对照品溶液在对照品溶液相应位置处无干扰,见图1。
线性关系考察:精密量取阿魏酸对照品溶液(质量浓度为0.069 7 g/L)1,2,3,5,8,10,12μL,进样,测定其峰面积,其对应的峰面积依次为341 822,692 317,1 043 048,1 728 576,2 800 213,3 497 125,4 199 254。以峰面积积分值(Y)对进样量(X)进行回归分析,得标准曲线方程 Y=5.03×106X-8 775,r=0.999 85(n=7)。结果表明,阿魏酸进样量在0.069 7~0.836 4μg范围内与峰面积线性关系良好。

图1 高效液相色谱图
精密度试验:精密吸取对照品溶液10μL,连续进样6次,测定峰面积分别为3 497 125,3 515 576,3 468 241,3 475 489,3 507 162,3 510 213。结果的 RSD为0.56%(n=6),表明仪器精密度良好。
稳定性试验:取同一供试品溶液,分别于0,2,5,7,10,12 h时进样测定峰面积。结果测得峰面积分别为674 032,669 425,674 971,659 269,667 543,661 931,其RSD为0.95%(n=6),表明供试品溶液在12 h内稳定。
重复性试验:取同一批(批号为151202)样品6份,依法制备供试品溶液,并按拟订色谱条件进样测定。结果阿魏酸平均含量为0.253mg/g,RSD为1.28%(n=6),表明方法重复性良好。
加样回收试验:精密量取已知含量的样品(批号为151202,质量浓度为0.013 28 g/L)6份,每份2 mL,置10 m L容量瓶中,分别加入对照品溶液(质量浓度为0.069 7 g/L)1.0,1.0,2.0,2.0,3.0,3.0m L,再加70%甲醇至刻度,摇匀,按拟订色谱条件进样测定,计算回收率。结果见表1。
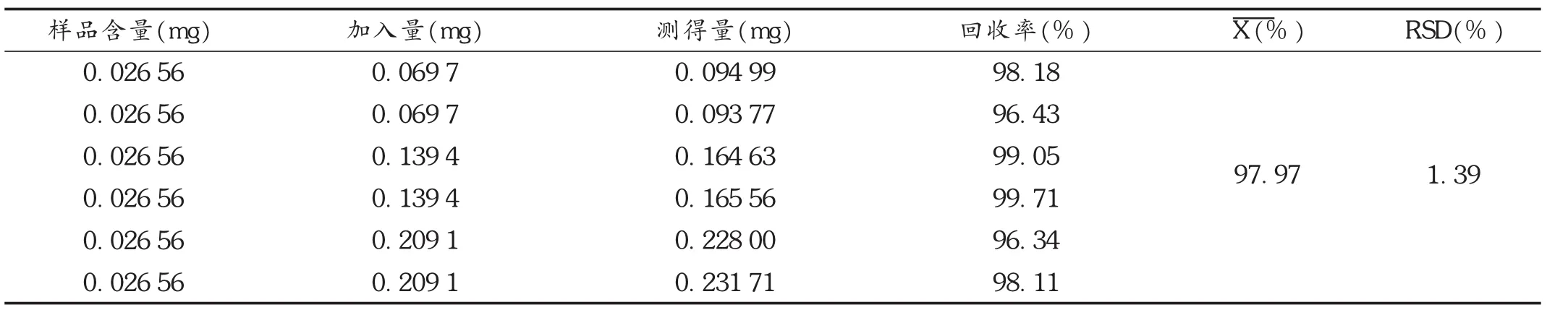
表1 阿魏酸加样回收试验结果(n=6)
2.4 样品含量测定
取2批(批号分别为151202,160210)样品,各2份,依法制备供试品溶液,分别精密吸取供试品溶液各10μL,进样,测定峰面积,记录色谱图,按外标法计算阿魏酸的含量。结果,批号为151202,160210样品中阿魏酸的含量分别为 0.253,0.256 mg/g和 0.261,0.259mg/g(n=2)。
3 讨论
3.1 测定波长选择[1]464
取对照品溶液和供试品溶液,用紫外分光光度计于200~400 nm波长范围内进行扫描,结果两者均于316 nm波长处有最大吸收峰,与光谱图一致,故选择316 nm作为阿魏酸含量测定的波长。
3.2 流动相选择
分别选择乙腈-0.085%磷酸溶液、甲醇-0.5%冰乙酸、甲醇-水作为流动相,经过多次有机相和无机相配比调整,最后确定色谱条件为0.085%磷酸溶液-乙腈(83∶17),在理论板数、分离度、峰形及出峰时间等方面均达到满意的结果。
3.3 提取溶剂筛选[9-13]
分别以温水、70%甲醇作为溶剂超声提取和密塞加热回流30min处理样品,由于阿魏酸结构的特点,采用水作为提取溶剂,导致其色谱峰拖尾、开叉或裂分,影响测定结果,色谱峰的理论板数大多低于4 000。而70%甲醇能完全溶解样品,且无拖尾现象,峰形对称,理论板数均大于5 000,故本方法采用70%甲醇为提取溶剂。
3.4 提取时间筛选[14-15]
曾考察加热回流20,30,40 min,含量测定结果表明,提取30,40 min的样品阿魏酸含量无明显差异,故确定提取时间为30min。
综上所述,采用高效液相色谱法以当归中阿魏酸作为指标性成分,对八珍颗粒中的当归成分进行有效的质量控制,简单易行,保留时间稳定,结果准确、可靠,可控制该药品的内在质量。
[1]国家药典委员会.中华人民共和国药典(一部)[M].北京:中国医药科技出版社,2015.
[2]寇俊萍,唐新娟,庄书斐,等.补益气血方剂对血液流变学的影响[J].中医药学报,2003,31(5):28.
[3]王 芳,李 东.当归的化学及药理研究进展[J].中国药房,2003,14(10):630.
[4]邓永健,郭志伟,王 萌.当归的化学成份及其药理作用研究进展[J].新疆中医药,2006,24(5):109.
[5]夏 泉,张 平,李绍平.当归的药理作用研究进展[J].时珍国医国药,2004,15(3):164.
[6]王中华.当归中阿魏酸含量测定及质量标准的研究[J].河北中医,2012,34(7):1058.
[7]赵子明,曹英莉.高效液相色谱法测定浓缩当归丸中阿魏酸含量[J].中国药业,2010,19(10):30.
[8]曾 宇,杜迎翔,马世平.反相高效液相色谱法测定当归芍药散中阿魏酸和芍药苷的含量[J].中国药科大学学报,2004,35(4):341.
[9]王映芬,何燕萍,蔡 阳.复方偏头痛冲剂主要成分当归川芎中阿魏酸的含量测定[J].北京大学学报:医学版,1994,26(3):218.
[10]李顺意,蔡 敏,韩凤梅.高效液相色谱法测定八珍益母颗粒中芍药苷和阿魏酸的含量[J].中国中药杂志,2001,26(9):631-632.
[11]刘晓芳,刘养清,赵 平.7种中药材中阿魏酸含量的测定[J].山西中医学院学报,2009,10(1):18-19.
[12]王晓飞,于 玲,葛海生,等.双波长反相高效液相色谱法测定柴辛鼻敏康颗粒中芍药苷和阿魏酸的含量[J].中国药业,2011,20(18):86.
[13]申 安.高效液相色谱法测定不同产地当归中阿魏酸的含量[J].中医学报,2015,30(3):421-422.
[14]黄 玮,郭 宁.HPLC法测定防栓胶囊中阿魏酸的含量[J].淮海医药杂志,2012,30(6):542-543.
[15]刘蓉梅,黄罗生.高效液相色谱法测定当归中阿魏酸含量方法的探讨[J].东南大学学报:医学版,2003,22(2):98-101.
Content Determ ination of Eru lic Acid in Bazhen G ranu les by RP-HPLC
Liu Haiyan
(Zhoukou institute for food and drug control,Zhoukou,Henan,China 466000)
Ob jective To establish an HPLC method for determining the content of ferulic acid in Bazhen Granules.M ethods Agilent Hc-C18column(250 mm×4.6 mm,5μm)chromatography column was used;the mobile phase was 0.085% phosphoric acid solutionacetonitrile(83∶17),flow rate 1.0 mL/min,detection wavelength 316 nm,column temperature 35℃.Resu lts The linear range of ferulic acid was in 0.069 7-0.836 4μg(r=0.999 85),the average recovery rate was 97.97%,RSD was 1.39%(n=6).Conclusion The method is reliable,accurate and reproducible,and can be used for the quality control of ferulic acid in Bazhen Granules.
RP-HPLC;Bazhen Granules;ferulic acid;content determination
R284.1;R286.0
A
1006-4931(2017)03-0027-03
2016-11-07)
10.3969/j.issn.1006-4931.2017.03.009
刘海燕,女,大学本科,副主任药师,主要从事中药、抗生素和微生物检验工作,(电子信箱)13939418667@163.com。
