无标准品时新精神活性物质甲卡西酮的定性定量检验
2017-04-10魏万里张绍雨刘景宁1
魏万里, 窦 莉, 张绍雨, 刘景宁1,
(1.南京工业大学, 江苏南京 210031; 2.无锡市公安局刑事科学技术研究所, 江苏无锡 214002;3.江苏警官学院, 江苏南京 210031)
无标准品时新精神活性物质甲卡西酮的定性定量检验
魏万里1,2, 窦 莉1,2, 张绍雨3, 刘景宁1,3
(1.南京工业大学, 江苏南京 210031; 2.无锡市公安局刑事科学技术研究所, 江苏无锡 214002;3.江苏警官学院, 江苏南京 210031)
目的 在无甲卡西酮标准品的情况下,对新精神活性物质甲卡西酮进行检验、鉴定。方法 可疑样品先经高效液相色谱- 飞行时间质谱(HPLC-TOF-MS)进行新精神活性物质筛选,通过精确分子量确定可疑新精神活性物质分子式,再经气相色谱- 质谱联用(GC-MS)及傅里叶变换衰减全反射红外光谱(ATR-FTIR)分析和文献值比对确定甲卡西酮的分子结构,最后用核磁共振氢谱(1H NMR)对甲卡西酮的分子结构进行确认并进行定量分析。结果 样品得到与甲卡西酮一致的分子式,且其质谱数据、红外数据均与甲卡西酮文献值一致,并经核磁共振氢谱证实,确认检材中含有甲卡西酮成分,测得其含量为24.6%。结论 本方法简单、准确、可靠,适用于无标准品时对新精神活性物质甲卡西酮进行检验、鉴定。
新精神活性物质; 甲卡西酮; 飞行时间质谱; 傅里叶变换红外光谱; 衰减全反射; 核磁共振氢谱; 核磁共振定量
0 引言
联合国毒品与犯罪问题办公室(UNODC)对“新精神活性物质”(New Psychoactive Substance, NPS)的定义是[1]:未被《1961年麻醉品单一公约》或《1971年精神药物公约》管制,但以单体纯净物或制剂的形式滥用,会对公众健康带来威胁的物质。这里的“新”并不一定代表是新的合成,有些NPS在四十多年前就首次合成了,但是,最近才成为市场上非法滥用的物质。UNODC发布的2015世界毒品问题报告中指出:目前全球九十多个国家和地区共发现、报告的“新精神活性物质”有9大类,共计541种。其中卡西酮类NPS中的甲卡西酮(Methcathinone,MC)是目前国际上和国内滥用较为严重的一种新精神活性物质。市场上所滥用的新精神活性物质,一般均是其盐类,以盐酸盐居多。如甲卡西酮,市场上以盐酸甲卡西酮(包括左旋体盐酸盐、右旋体盐酸盐)形式滥用,本研究中的可疑样品即是甲卡西酮盐酸盐,在此不再特别标注盐酸盐。
新精神活性物质的品种正在不断增加,滥用日趋严重,为依法打击新精神活性物质的违法犯罪活动,必须对新精神活性物质进行定性定量检验,做出科学正确的鉴定结论。目前,国内关于新精神活性物质检验的方法、标准几近空白,新精神活性物质的定性定量标准品也仅有十多种,无法满足基层毒品检验部门办案需要。国内已有的几种毒品检验国家标准均是采用气相色谱或液相色谱与质谱联用技术[2-4],与毒品标准品进行比对,进行定性定量检验。
国内外有关新精神活性物质检验的研究报道尚少,尤其鲜见核磁共振(NMR)技术用于NPS定量分析的报道。究其原因,主要是获取NPS标准品十分困难。NMR作为未知物定性的最有力的手段在国外已经广泛运用于NPS检测中[2-5],国内也有学者尝试运用NMR技术鉴定未知NPS[6、7];但由于核磁共振设备价格昂贵,难以在基层毒品检测部门普及。随着科技的发展,价格低廉的台式核磁共振仪,其分辨率及灵敏度已经可以满足NPS定性、定量检验的要求。核磁共振定量技术已经成为天然产物分析[8]、体内药物代谢物分析[9]、药品基准物质测定[10]的主要手段;NMR定量具有快速、简单、准确、专属性强等特点,应用前景广阔。
本文综合运用HPLC-TOF-MS、GC-MS、ATR-FTIR、NMR技术,研究在缺少标准品的情况下,对涉及甲卡西酮案件中的样品进行定性定量分析的检验方法。
1 实验部分
1.1 仪器
安捷伦1260系列液相色谱、6230飞行时间液质联用仪, MassHunter工作站(Version B.05.00);Nicolet iN10傅里叶变换显微红外光谱仪,附带Nicolet iZ10 FT-IR模块及ATR附件(美国ThermoFisher公司) ;TRACE GC ULTRA/ISQ气相色谱质谱联用仪(美国 ThermoFisher公司) ;picoSpin 80核磁共振波谱仪(美国ThermoFisher公司)。
1.2 试剂
纯水(Optima高纯级,美国Fisher);环己烷(ACS/HPLC级,J&K百灵威);丙酮(LC级,德国Merck); 咖啡因标准品(99.8%,购自中国标准物质采购中心);甲醇、乙腈、丙酮、乙醚均为HPLC级试剂;三甲基硅基丙酸盐TMSP-2,2,3,3-d4(D,98%,J&K百灵威);重水(D,99.8%,J&K百灵威)。
1.3 HPLC-TOF-MS分析条件
HPLC条件 色谱柱:ZORBAX Eclipse XDB-C18柱, 4.6 mm×100 mm,1.8 μm;柱温: 60 ℃;流动相A: 纯水,B: 乙腈;流速: 0.3 mL/min;梯度洗脱:开始30% B;12.0 min,90% B;15.0 min 30% B;分析时间: 15.0 min;后运行时间: 3.0 min;总分析时间: 18.0 min;进样体积: 10 μL。
TOF条件 安捷伦DualAJS ESI离子源;正离子模式;干燥气温度和流速: 320 ℃, 8 L/min;鞘流气温度和流速: 380 ℃, 12 L/min;雾化器压力: 27 psi;喷嘴电压: 500 V;毛细管电压: 3 750 V;碰撞电压: 150 V;参比离子(m/z): 121.050 9和922.009 8, 分析时连续恒流使用参比A;质量范围:m/z 50~1 050;扫描速度:3 Hz;其它操作参数都基于标准的质谱自动调谐和质量轴校正。
1.4 GC-MS分析条件
色谱条件 色谱柱: DB-5 ms(30 m×0.25 mm×0.25 μm);柱温:80 ℃(1 min)20 ℃/min 280 ℃(10min),进样口: 250 ℃,进样量:1.0 μL,分流进样:分流比100∶1;载气:氦气(纯度>99.999%);流速:1.0 mL/min。
质谱条件 传输线温:280 ℃;离子源温度:230 ℃;电离模式:EI;轰击能量:70 eV;灯丝电流:30 μA;扫描范围:40~500 amu;扫描模式:SCAN。
1.5 ATR-FTIR分析条件
扫描范围: 4 000 cm-1~400 cm-1,扫描次数32次,分辨率:4 cm-1,测试方式:ATR,检测器:DTGS。
1.61H NMR分析条件
90°射频发射脉冲60 μs、采样时间1 000 ms和弛豫延迟时间15 s。核磁谱图的累加次数为20~100次不等。核磁数据处理:采用MestReNova10.0版进行数据处理。所有核磁谱图数据处理方法一致: 充零至64 k点,向前线性和向后线性预测,手动校正零级相位(PH0);窗函数为指数函数(0.60 Hz)。
1.7 样品准备
可疑样品用甲醇溶解配制成1.0 mg/mL溶液供GC-MS测试;可疑样品用流动相溶解配制成1.0 mg/mL溶液,再用流动相稀释至100.0 μg/mL、10.0 μg/mL,过0.22 μm滤膜后供HPLC-TOF-MS测试。准确称取适量样品溶于重水,加适量TMSP后供NMR测试。
2 结果与讨论
2.1 自建NPS精确分子量数据库,通过HPLC-TOF-MS分析筛选可疑的NPS样品
根据UNODC发布列出的NPS品种表,用MassHunter工作站软件编制了包括所有NPS品种的单一同位素精确分子量数据库,其中一些NPS属于同分异构体,有相同的单一同位素精确分子量,筛选时应注意。
可疑样品进样100 ng, 进行HPLC-TOF-MS分析。使用MassHunter 定性分析软件处理上述得到的数据文件,采用“分子式查询(Find by Formula)”方式,调用前面建立的NPS精确分子量数据库,根据精确质量数,同位素丰度,同位素间隔匹配的综合分值,得到最匹配的可疑NPS分子式。结果显示样品中可能含有甲卡西酮成分,见图1a。同时使用自建药物库筛选,得到可能含有咖啡因成分,见图1b。图1为样品的提取化合物叠加色谱图。
“分子式查询”数据挖掘算法,可以调用数据库中的所有分子式逐个自动检索TOF数据文件,以确认是否存在该化合物,确保筛选时已知的NPS一个也不遗留。
2.2 GC-MS及FT-ATR-IR分析确定分子结构
将可疑样品进行GC-MS分析,结果显示样品的主要成分为甲卡西酮、咖啡因及对苯二酚。见图2,图中1为对苯二酚、2为甲卡西酮、3为咖啡因,其中对苯二酚上的2个羟基造成了非极性色谱柱上色谱峰的拖尾现象。对咖啡因、对苯二酚,用标准样品进行了确认。疑是甲卡西酮的成分,其质谱检索结果显示与SWGDRUG质谱库2.4版[11]中的甲卡西酮相一致,见图3,图3a为样品中疑是甲卡西酮成分的质谱图,图3b为SWGDRUG质谱库中甲卡西酮的质谱图。

图2 样品的GC-MS总离子流色谱图
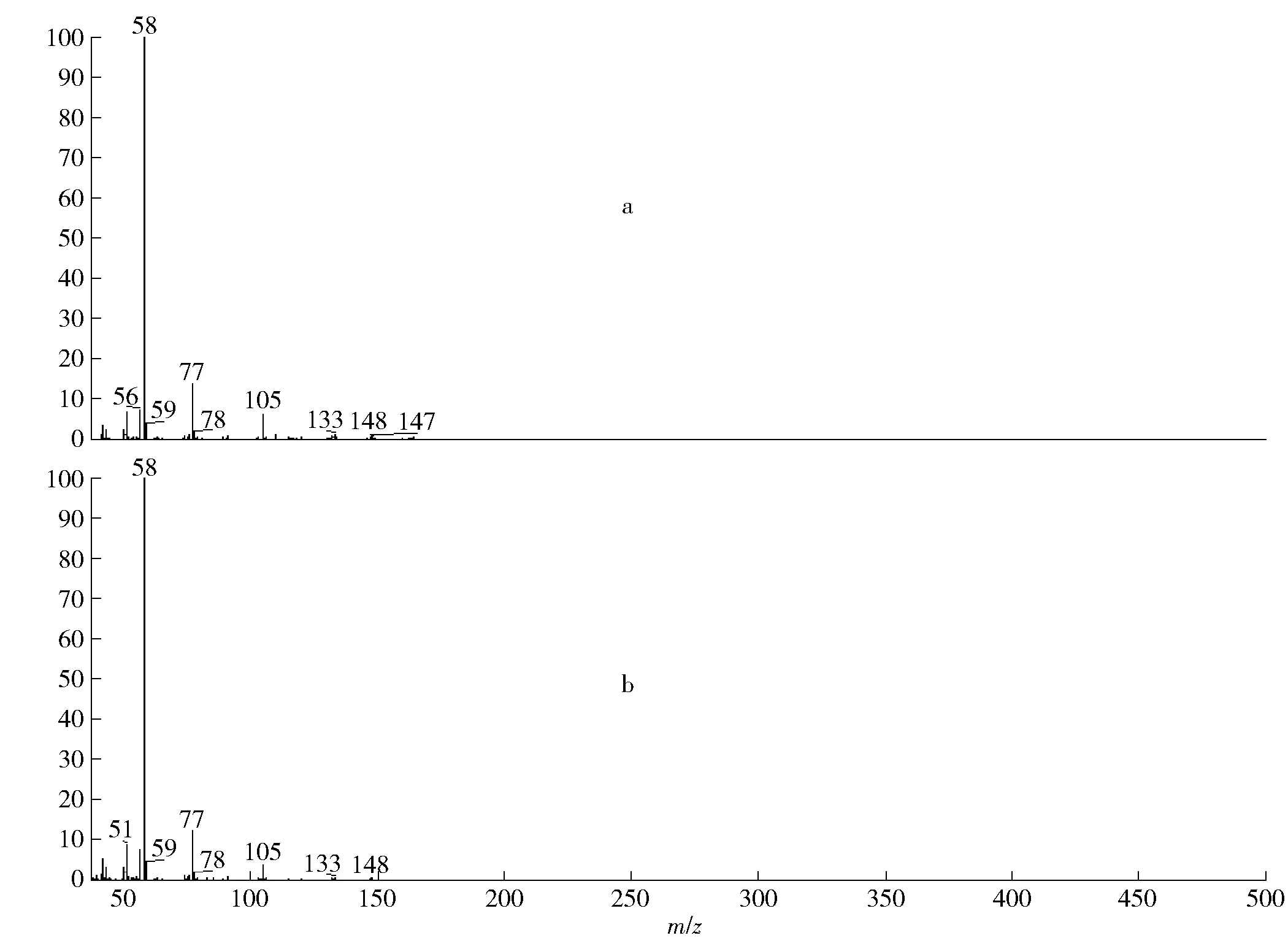
图3 样品中疑似甲卡西酮成分与SWGDRUG数据库中甲卡西酮质谱比对
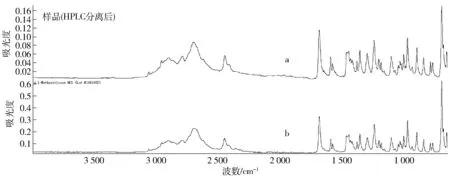
图4 样品中疑似甲卡西酮成分红外光谱与SWGDRUG数据库中甲卡西酮红外光谱的比对
为得到纯净的可疑NPS,将进样量提高到1 μg,根据保留时间收集HPLC-TOF-MS分析时疑是甲卡西酮的馏分(此时不接TOF-MS),集中5次分析的馏分,加入10 μL氯化氢乙醇溶液,60 ℃下氮吹挥干,丙酮溶解后,滴加在ATR上,待溶剂挥干后,采集IR数据,得到该可疑NPS的红外光谱,检索发现与SWDRUG提供的NPS红外谱库1.2版[12]中的盐酸甲卡西酮相一致,见图4。图4a为样品中疑是甲卡西酮的红外谱图,图4b为SWDRUG提供的盐酸甲卡西酮的红外谱图。
TOF筛选结果只说明与可疑的NPS分子式一致,其是否与可疑NPS具有一致的分子结构,再需要通过质谱、红外数据确定。虽然没有标准品比对,但可以和文献值比对,包括权威网站提供的数据,如SWGDRUG质谱库2.4版(包括2 372种化合物)和SWGDRUG红外谱库1.2版(包括216化合物),经过比对我们可以确定该化合物的结构,去证实是否属于NPS。
根据SWGDRUG的建议[13],质谱、红外、核磁共振检测均属于A类定性技术,2个A类技术联用,已经足以对查获的毒品进行正确的定性。假如文献中只能找到质谱或红外数据,那就还需要其他技术辅助才能正确定性。
2.31H NMR确证分子结构
由上述分析测试得知,样品主要成分是甲卡西酮、咖啡因及对苯二酚,根据它们在酸碱水溶液中的溶解度不同,取0.2 g样品溶于5 mL纯水中,调节pH值为10,用2 mL乙醚提取甲卡西酮,弃去水层,再加5 mL纯水调pH值为2,提取盐酸甲卡西酮,弃去有机层。重复上述过程3次,最后的乙醚溶液中加入100 μL氯化氢乙醇溶液,氮吹60℃挥干得到少量的甲卡西酮粗制品,500 μL重水溶解,加适量TMSP,用picoSpin台式核磁共振仪做核磁共振氢谱分析。
样品的核磁共振氢谱见图5,发现与甲卡西酮相吻合。根据核磁共振谱图,确认核磁谱图中的各个峰是否与结构相符,也可以与文献的核磁共振图谱比对。MestReNova10.0软件可以根据分子结构预测该结构的核磁共振氢谱,虽然预测的谱图与真实谱图有差异,但大体峰型比例是基本一致的,可以作为分析NMR谱图的参考。由此进一步确认该样品中是否含有甲卡西酮成分。
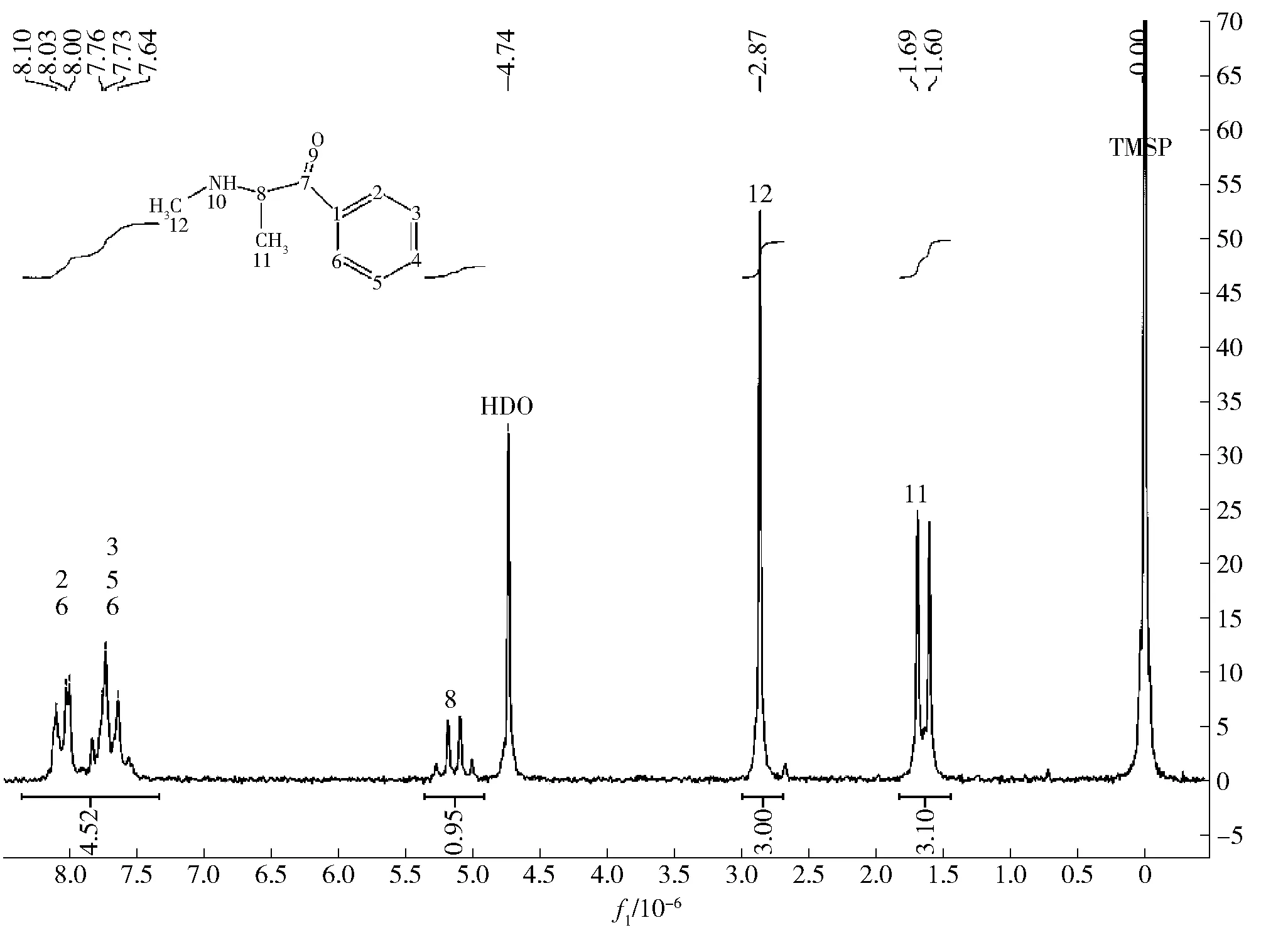
图5 样品的1H NMR图谱确证甲卡西酮(归属和原子核数确定)
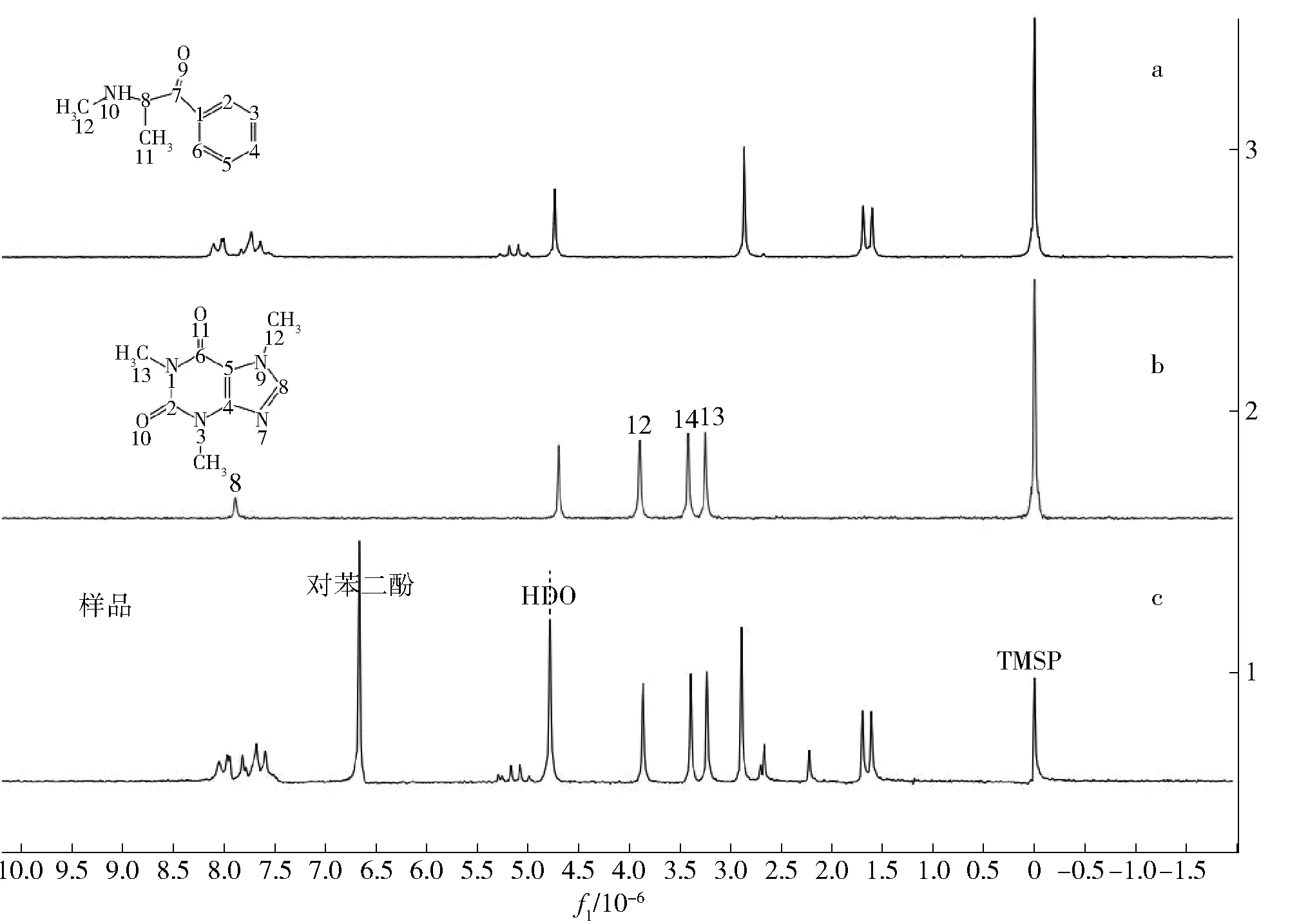
图6 甲卡西酮、咖啡因与样品1H NMR 图谱对比
样品核磁共振谱图简单时,可以不经提纯,直接从原始样品的核磁共振谱中得到足够的信息,确认是否含有该NPS。图6c为原始样品的核磁共振氢谱,该谱图基本就是上述3种主要成分氢谱的叠加,图6a为甲卡西酮的1H NMR图谱,图6b为咖啡因的1H NMR图谱。因此,核磁共振氢谱也证实了该样品的3种主要成分为甲卡西酮、咖啡因和对苯二酚。
2.41HNMR定量分析
核磁共振定量的原理,核磁共振谱峰信号的峰面积或强度,在一定的实验条件下,谱峰面积或强度正比于引起此信号的质子数。picoSpinTM80核磁共振波谱仪射频线圈内的有效容积约为70 nL,是一个定值,则有下列公式:A=K×N×M(公式1),M为此物质的摩尔浓度(为计算方便,本文采用mmol/L计量),N为该谱峰的氢原子核数,A为该谱峰面积(仪器绝对积分值),K为谱峰面积与摩尔浓度之间的换算因子。在特定的条件下,K是一个常数,与射频线圈内的有效容积及核磁共振测试参数有关。以纯水、色谱纯丙酮、色谱纯环己烷3种液体与咖啡因固体,取合适位置的氢,分别测得峰面积,均测5次,取平均峰面积,根据上述公式,计算得到换算因子K值,见表1。纯液体以36 ℃(射频线圈内的温度)的密度换算为摩尔浓度,咖啡因在36 ℃环境下通过称重溶于重水中配得已知浓度。实验结果显示,4种物质测得的K值基本一致,换算因子K的相对标准偏差(RSD)为0.70%,取4种物质的平均K值5.800 8为该条件下的换算因子,去计算待测化合物的未知浓度。
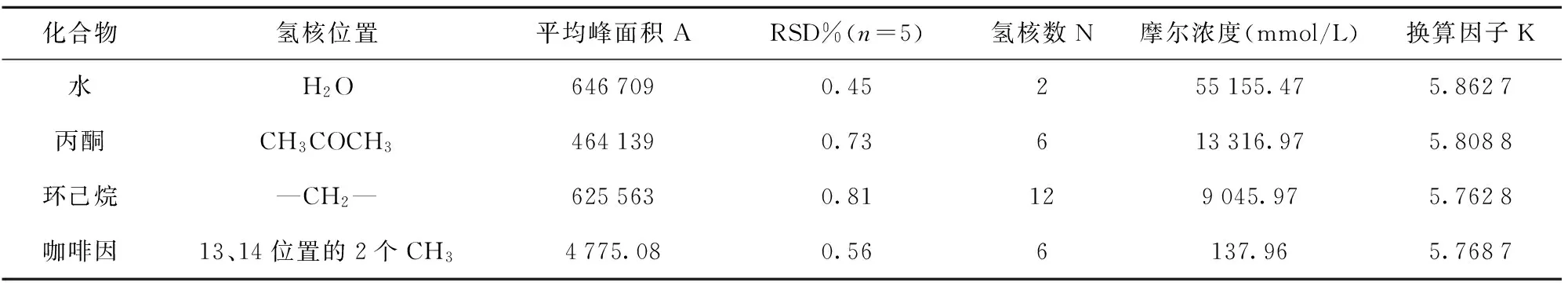
表1 4种不同化合物测得的换算因子K值
同样操作,测得了样品中甲卡西酮、咖啡因、对苯二酚的摩尔浓度,换算成百分浓度,见表2,3种主要成分的总含量为95.5%。采用食品安全国家标准GB 5009.139—2014(饮料中咖啡因的测定)中的HPLC法测得样品中咖啡因的浓度为21.4%(n=5, RSD=1.3%)。核磁共振法的结果与HPLC法比较,核磁共振法的测试结果在国标HPLC法允许的标准偏差范围内,表明核磁共振法的测试结果是可靠的。

表2 qNMR法测得的样品中3种主要成分的含量
注:准确称取705.0 mg样品,重水溶解定容至10.0 mL(36 ℃);24.6%为甲卡西酮的含量,换算成盐酸甲卡西酮应为30.1%。
3 结论
本文建立了一种在无标准品时,用飞行时间质谱筛选甲卡西酮,再根据质谱和红外光谱数据与文献值比对确定分子结构,最后用核磁共振氢谱验证甲卡西酮分子结构和对甲卡西酮进行定量分析的方法。该方法适用于无甲卡西酮标准品时,对可疑的甲卡西酮样品进行检验鉴定。该方法表明,在没有NPS标准品时,也能对NPS进行准确的定性定量分析,为依法打击新精神活性物质违法犯罪活动提供科学的证据。
[1] UNITED NATIONS OFFICE ON DRUGS AND CRIME.What are NPS[EB/OL].https:∥www.unodc.org/LSS/Page/NPS,2016-4-15.
[2] PEDERSEN A,REITZEL L,JOHANSEN S,et al.In vitro metabolism studies on mephedrone and analysis of forensic cases[J].Drug testing and analysis,2013,5(6):430-438.
[3] AMATO J,IACCARINO N,PAGANO B,et al.NMR Assignment of N-(1-adamantyl)-1-pentyl-1H-indazole-3-carboxamide Seized as Herbal Incense for the First Time
in Italy[J].Journal of Forensic Science & Criminology,2014,2(1):1-6.
[4] UCHIYAMA N,ASAKAWA K,KIKURAHANAJIRI R,et al.A new pyrazole-carboxamide type synthetic cannabinoid AB-CHFUPYCA [N-(1-amino-3-methyl-1-oxobutan-2-yl)-1-(cyclohexylmethyl)-3-(4-fluorophenyl)-1H-pyrazole-5-carboxamide] identified in illegal products[J].Forensic Toxicology,2015,33(2):367-373.
[5] SHEVYRIN V,MELKOZEROV V,NEVERO A,et al.Identification and analytical characteristics of synthetic cannabinoids with an indazole-3-carboxamide structure bearing a N-1-methoxycarbonylalkyl group[J].Analytical and Bioanalytical Chemistry,2015,407(21):6301-6315.
[6] 黄星,王蔚昕,徐鹏,等. N-甲基-3,4-亚甲二氧基卡西酮的确证[J].中国法医学杂志,2012,27(6):474-476.
[7] 曹芳琦,李茂盛,袁晓亮,等. 4-甲基乙卡西酮的鉴定研究[J].中国司法鉴定,2015,80(3):43-46.
[8] PAULI G,GODECKE T,JAKI B,et al.Quantitative 1H NMR: Development and Potential of an Analytical Method-an Update[J].Journal of Natural Products,2012,75 (4):834-851.
[9] LERCHE M,MEIER S,JENSEN P,et al.Quantitative dynamic nuclear polarization-NMR on blood plasma for assays of drug metabolism[J].NMR in Biomedicine,2011,24(1):96-103.
[10] WU Y, HE Y,HE W,et al.Application of quantitative 1H NMR for the calibration of protoberberine alkaloid reference standards[J].Journal of Pharmaceutical and Biomedical Analysis,2014,90:92-97.
[11] SWGDRUG. SWGDRUG MS Library Version 2.4 [EB/OL].http:∥www.swgdrug.org/ms.htm,2015-12-30/2016-04-15.
[12] SWGDRUG.SWGDRUG IR Library Version 1.2 [EB/OL].http:∥www.swgdrug.org/ir.htm,2015-05-26/2016-04-15.
[13] SWGDRUG.SWGDRUG Recommendations Edition 7.0[EB/OL]. http:∥www.swgdrug.org/approved.htm,2014-08-14/2016-4-15.
(责任编辑 于瑞华)
江苏省第五期“333工程”科研项目(BRA2016101)。
魏万里(1972—),男,江苏无锡人,高级工程师。主要研究方向为毒物、毒品、微量物证的检验鉴定。
D918.93
