COG6基因新突变致新生儿期起病的先天性糖基化障碍1例并文献复习
2017-04-07吴冰冰李宛星王慧君周文浩
吴冰冰 李宛星 杨 琳 王慧君 周文浩
·论著·
COG6基因新突变致新生儿期起病的先天性糖基化障碍1例并文献复习
吴冰冰1李宛星1杨 琳 王慧君 周文浩
目的 诊断1例COG6基因复合杂合突变所致的先天性糖基化障碍(CDG),为CDG患儿的的早期诊断、制定干预措施和结局预测提供依据。方法 总结1例携带有COG6复合杂合突变的CDG患儿的临床表型、家系sanger验证信息、影像学表现、实验室检查和随访信息,对COG6及其他Golgi复合体(COG)基因突变所致CDG的疾病表型行文献复习。结果 患儿因早产、生后反复气促吐沫1月余就诊,主要表现为不明原因反复高热伴肝酶异常,皮肤少汗,异常面容,并存在心、肺、肾、凝血和神经系统异常。行核心家系全外显子组检测发现COG6基因复合杂合突变c.511C>T (p.R171X)和c.540G>A (p.E180E),c.511C>T 来源于母亲,是人类基因突变数据库(HGMD)已报道的CDGⅡ型的致病突变;c.540G>A 来源于父亲,为新发突变。汇总专业版HGMD已报道的COG6-CDG 患儿9例加本文1例共10例表型(CDGⅡ型),异常面容,可表现为肝、皮肤、心脏、肾脏、骨骼、关节、凝血、免疫、神经系、听力和视觉异常或其他畸形等,多数患儿生长发育迟缓,预后不良,5例病死,存活者均进展为严重肝功能障碍伴反复感染。比COG-CDG其他亚型,临床表现更丰富、病情偏重且预后差。结论 新生儿期表现为不明原因高热伴肝酶异常,皮肤少汗,肌张力异常,或存在心、肾、免疫和凝血等多器官和系统功能异常的患儿,应高度怀疑COG6-CDG,此类患儿多数生长发育迟缓,预后不良,新生儿期通过基因测序可早期诊断。
COG6基因; 先天性糖基化障碍; 少汗型外胚层发育不良; 突变; 高尔基复合体
1 病例资料
患儿男,1月16 d,因“早产,生后反复气促吐沫1月余”于2016年4月至复旦大学附属儿科医院(我院)就诊。
患儿系G1P1,试管婴儿,孕周34周,因宫内窘迫行剖宫产,出生体重1 700 g。父母否认近亲结婚。
患儿于出生后因“窒息”入住外院(Apgar不详),诊断为新生儿窒息,新生儿呼吸窘迫综合征,呼吸衰竭,新生儿肺炎,早产儿,先天性心脏病,住院期间予肺表面活性物质替代、机械通气支持、抗生素、抗真菌、强心、利尿等治疗1月余,患儿仍反复气促、发热,转我院。
查体:呼吸急促、三凹征阳性,可闻及痰鸣音;肌张力偏高,未发现惊厥发作。入院第1 d血培养副血链球菌阳性;生化检查示肝酶异常(AST 164 IU·L-1,ALT 521 IU·L-1, γ-GT 243 IU·L-1);X线胸片示两肺渗出多;心脏彩超示先天性心脏病(室间隔缺损、卵圆孔未闭、肺动脉高压、永存左上腔静脉、心动过缓)。入院后予以常规抗生素抗感染治疗。患儿于住院第5 d起出现反复弛张热,体温最高39.5℃,经对症治疗后体温连续正常不超过4 d再发热。病程中患儿存在肺部感染(图1A),但无明显全身感染中毒症状,入院第8 d血培养复查转阴,此后多次痰、尿、脑脊液和血培养均阴性,CRP在正常范围,且抗生素治疗效果不佳,提示患儿发热不能用单纯肺部炎症解释。入院后肝酶持续异常,应用保肝药后复查肝酶仍高(AST 107 IU·L-1,ALT 571 IU·L-1,γ-GT 154 IU·L-1)。因患儿住院期间不明原因反复发热,于入院52 d在我院进行疑难病例讨论,患儿反复发热,结合查体皮肤光滑、皮纹少,平素出汗少,发热及热退时无明显出汗表现,裸露皮肤后发热可缓解,考虑外胚层发育不良可能。建议行皮肤病理活检,汗腺结构正常且发汗试验阳性,少汗型外胚层发育不良可能性大。头颅MRI示双侧脑室略饱满,后角少许陈旧性出血可能(图1B);B超检查示左肾内多发实质性小占位(图1C);头颅CT示下颌骨形态稍小,第二磨牙未见钙沉着落后于足月新生儿(图1D);腹部X线片示肠腔大量积气,X线胸片示新生儿支气管肺发育不良;入院2月余内分泌检查TSH升高(12.73 μlU·mL-1),考虑甲状腺功能减低,不能排除为早产儿暂时性甲状腺功能减低;血常规提示嗜酸性粒细胞缺乏。患儿住院治疗70 d后家属签字出院,出院时患儿仍处高流量吸氧,气促明显,需鼻饲喂养。出院诊断:发热(外胚层发热不良可能大),新生儿支气管肺发育不良,支气管肺炎,肝功能异常,先天性心脏病,心功能不全,贫血,低出生体重儿。出院后通讯随访得知患儿死亡,死亡时间、原因不详。

图1 患儿的影像学表现
注 A:X线胸片,B:头颅MRI, C:腹部B超,D头颅CT
为了明确患儿病因,于入院38 d时采集患儿及其父母的外周血各2 mL,应用高通量全血磁珠纯化试剂盒提取DNA,采用Illumina HiSeq X平台测序,与人类参考基因组序列比对,表明95%的目标捕获区域测序深度>50×。用WuXi NextCODE公司的临床序列分析软件(Clinic Sequence Analyzer,CSA)分析测序结果,发现患儿存在COG6突变,进行sanger验证,并提取父母DNA做家系sanger验证。图2显示患儿有COG6基因的复合杂合变异c.511C>T(p.R171X)和c.540G>A(p.E180E),c.511C>T来自患儿母亲,c.540G>A来自患儿父亲。c.511C>T是人类基因突变数据库(HGMD)已报道的先天性糖基化障碍(CDG)Ⅱ型的致病突变[1],无义突变导致蛋白质在171位翻译提前终止。c.540G>A未在HGMD报道,也未在千人基因组计划、ExAC数据集及我院分子诊断中心内部数据库中检索到,为罕见变异。虽为同义变异,但位于5号外显子最后一个碱基,在线预测软件MutationTaster也预测其影响剪接,为致病突变。结合患儿临床资料及基因测序结果,患儿诊断为COG6-CDG。
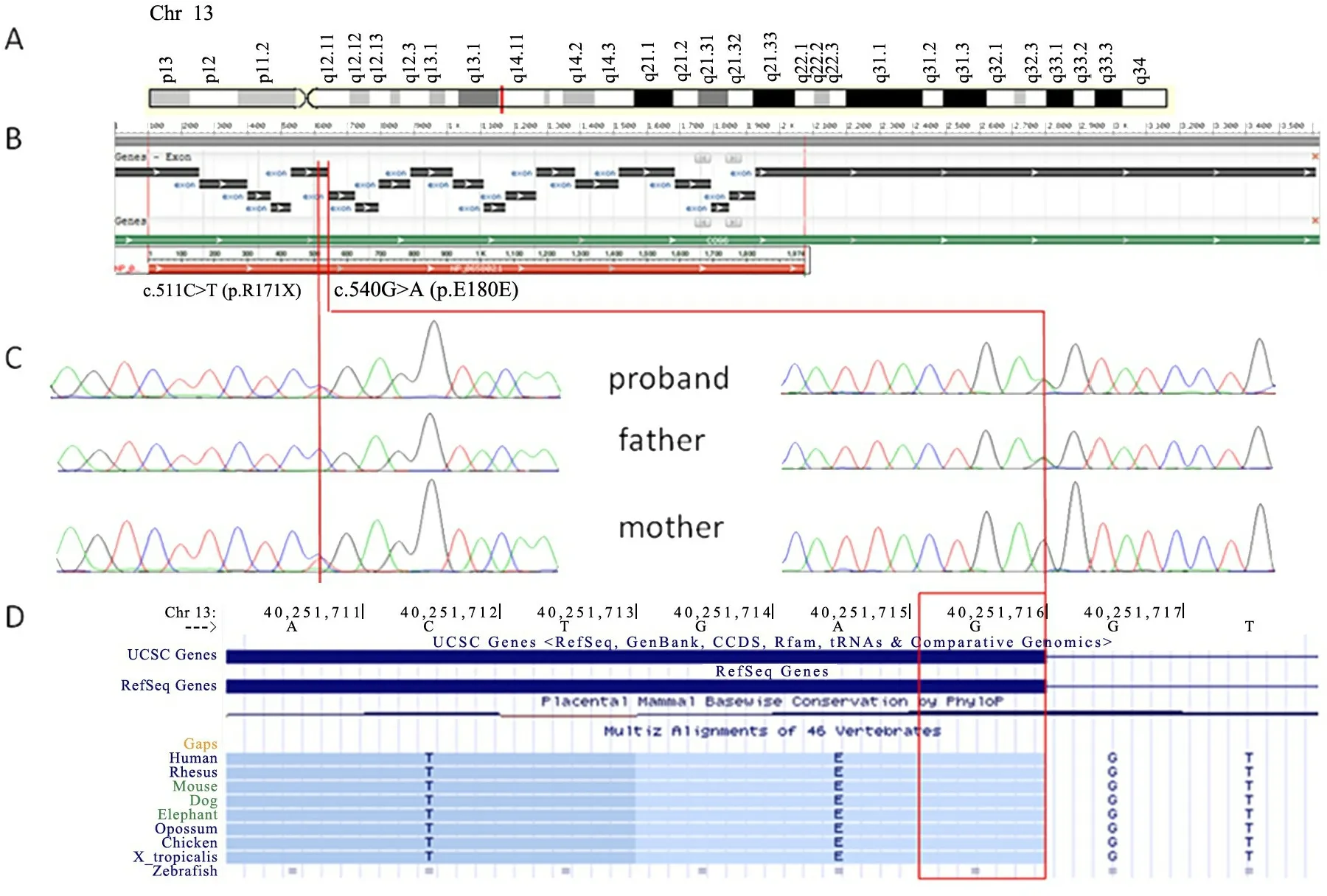
图2 患儿及其父母COG6基因检测
2 讨论
糖基化修饰是一种重要的转录后修饰,约70%的蛋白质在内质网和高尔基体被加上特定的糖链,糖蛋白在蛋白折叠、分子识别和黏附等过程中发挥重要作用[2]。CDG是一种罕见的先天性遗传代谢性疾病,由于蛋白质和脂质的糖基化过程无处不在,所以CDG 患儿均表现为累及多系统的功能障碍。截止2017年1月11日专业版HGMD已报道的CDG相关基因共39个,Golgi复合体(COG)基因突变所致CDG均为Ⅱ型,该型为发生于内质网和COG内的N-糖链加工修饰障碍[3]。COG有A、B两叶,COG1~4构成A叶,COG5~8构成B叶。目前已经发现了COG-1、-2、-4、-5、-6、-7和-8基因突变导致的CDG[2,4~8]。

本文患儿早产,因反复气促吐沫1月余入住我院。入院第5 d起,患儿出现反复弛张热,结合临床查体、皮肤活检结果并排除感染原因后,考虑少汗型外胚层发育不良可能大。少汗型外胚层发育不良是由于汗腺缺乏或汗腺功能异常导致的少汗和高热,还可同时存在皮肤、头发、指甲、牙齿的异常。既往报道病例中有8例患儿表现为外胚层发育不全,其中5例患儿生后即反复高热,排除各种病原体感染因素后考虑是由于患儿异常少汗所致,另外在疾病后期患儿由于肝功能异常导致的出血倾向也是反复高热的原因之一。Shaheen等[11]报道的患儿也表现为高热、皮肤少汗和角化过度,但其皮肤活检显示汗腺数量和结构正常,故推断其皮肤少汗可能是由于汗腺功能异常所致。分布于汗腺的M3胆碱受体作为GPCR家族蛋白高度糖基化,其糖基化障碍致可导致受体功能不全,从而影响汗腺功能[12]。本文患儿住院期间肝酶持续偏高,应用保肝药物后肝功能并无好转,这与既往国外报道的病例表型相符。肝细胞具有活跃的蛋白质合成和分泌功能,推测由于糖基化障碍造成不成熟蛋白质在肝胆系统的合成运输障碍而导致患儿的肝细胞损伤。另外,Lecca等[13]发现CDG患儿来源的纤维母细胞比正常对照组分泌更多的细胞外基质包括胶原蛋白等,这也可能是CDG患儿进展为小结节性肝硬化的原因之一。本文患儿左肾B超示左肾内多发实质小占位,结晶可能;既往报道4例肾脏异常中有1例存在右肾高回声,结合本例患儿表型推测CDG可致肾脏结石形成。

表1 已报道的9例及本文1例COG6-CDG患儿的临床表型
既往报道COG6-CDG病例中小头畸形7例,多数患儿并非出生后即表现为小头畸形,而是生长发育过程中脑发育迟缓所致。研究发现糖基化障碍会影响脑发育过程中神经元的迁移和突触形成[14,15],推测可能是导致患儿小头畸形的原因之一。本文患儿表现为小下颌和牙齿发育落后于足月新生儿,但并未发现小头畸形,与本文患儿年龄尚小、临床表型尚未充分表现有关。糖免疫学研究揭示了糖基化蛋白在免疫细胞的成熟、抗原抗体的相互识别中发挥重要的作用,从而参与调控固有免疫和适应性免疫[16,17]。既往报道中6例患儿在病程后期存在反复感染,4例存在免疫异常。本文患儿诊断为支气管肺炎,X线胸片示两肺渗出多、右上肺段实变不张,腹部B超显示肠腔大量积气且不能耐受喂养提示胃肠道可能存在异常。可能由于患儿年龄尚小、疾病并未进展到后期,所以表型并不典型。
综上所述,本文患儿与既往报道COG6-CDG病例的临床表型高度相符,但由于本文患儿年龄小,有些临床表型尚未出现。

表2 COG-CDG各亚型临床表型总结与比较
注 1)反复的腹泻和肺部感染
3 结论
由于目前国外已报道的COG6基因突变所致的CDG患儿只有9例。本文通过核心家系全外显子组检测明确诊断1例COG6-CDG患儿,补充和丰富了该疾病的临床表型谱和基因突变谱。新生儿期表现为不明原因高热伴肝酶异常,皮肤少汗,肌张力异常,或累及心脏、肾脏、免疫、凝血等多系统的功能异常的患儿,应高度怀疑COG6-CDG,多数患儿生长发育迟缓、预后不良。新生儿期通过基因测序可早期诊断该病,为患儿制定早期干预治疗措施提供依据。
[1]Rymen D,Keldermans L,Race V,et al.COG5-CDG:expanding the clinical spectrum.Orphanet J Rare Dis,2012,7:94
[2]Freeze HH,Chong JX,Bamshad MJ,et al.Solving glycosylation disorders:fundamental approaches reveal complicated pathways.Am J Hum Genet,2014,94(2):161-175
[3]Rymen D,Winter J,Van Hasselt PM,et al.Key features and clinical variability of COG6-CDG.Mol Genet Metab,2015,116(3):163-170
[4]Lubbehusen J,Thiel C,Rind N,et al.Fatal outcome due to deficiency of subunit 6 of the conserved oligomeric Golgi complex leading to a new type of congenital disorders of glycosylation.Hum Mol Genet,2010,19(18):3623-3633
[5]Huybrechts S,De Laet C,Bontems P,et al.Deficiency of Subunit 6 of the Conserved Oligomeric Golgi Complex (COG6-CDG):Second Patient,Different Phenotype.JIMD Rep,2012,4:103-108
[6]Shaheen R,Ansari S,Alshammari MJ,et al.A novel syndrome of hypohidrosis and intellectual disability is linked to COG6 deficiency.J Med Genet,2013,50(7):431-436
[7]Rymen D,Jaeken J.Skin manifestations in CDG.J Inherit Metab Dis,2014,37(5):699-708
[8]Lecca MR,Maag C,Berger EG,et al.Fibrotic response in fibroblasts from congenital disorders of glycosylation.J Cell Mol Med,2011,15(8):1788-1796
[9]Vuillaumier-Barrot S,Bouchet-Seraphin C,Chelbi M,et al.Identification of mutations in TMEM5 and ISPD as a cause of severe cobblestone lissencephaly.Am J Hum Genet,2012,91(6):1135-1143
[10]Dityatev A,Dityateva G,Sytnyk V,et al.Polysialylated neural cell adhesion molecule promotes remodeling and formation of hippocampal synapses.J Neurosci,2004,24(42):9372-9382
[11]van Kooyk Y,Rabinovich GA.Protein-glycan interactions in the control of innate and adaptive immune responses.Nat Immunol,2008,9(6):593-601
[12]Monticelli M,Ferro T,Jaeken J,et al.Immunological aspects of congenital disorders of glycosylation (CDG):a review.J Inherit Metab Dis,2016,39(6):765-780
[13]朱丽,张蓉,张淑莲,等.中国不同胎龄新生儿出生体重曲线研制.中华儿科杂志,2015,53(2):97-103.
[14] Zeevaert R,Foulquier F,Dimitrov B,et al.Cerebrocostomandibular-like syndrome and a mutation in the conserved oligomeric Golgi complex,subunit 1.Hum Mol Genet,2009,18(3):517-524.
[15] Kodera H,Ando N,Yuasa I,et al.Mutations in COG2 encoding a subunit of the conserved oligomeric golgi complex cause a congenital disorder of glycosylation.Clin Genet,2015,87(5):455-460.
[16]Reynders E,Foulquier F,Leao TE,et al.Golgi function and dysfunction in the first COG4-deficient CDG type II patient.Hum Mol Genet,2009,18(17):3244-3256
[17]Wu X,Steet RA,Bohorov O,et al.Mutation of the COG complex subunit gene COG7 causes a lethal congenital disorder.Nat Med,2004,10(5):518-523.
[18]Foulquier F,Ungar D,Reynders E,et al.A new inborn error of glycosylation due to a Cog8 deficiency reveals a critical role for the Cog1-Cog8 interaction in COG complex formation.Hum Mol Genet,2007,16(7):717-730
[19]Morava E,Zeevaert R,Korsch E,et al.A common mutation in the COG7 gene with a consistent phenotype including microcephaly,adducted thumbs,growth retardation,VSD and episodes of hyperthermia.Eur J Hum Genet,2007,15(6):638-645
(本文编辑:张崇凡,孙晋枫)
A case of neonatal congenital disorders of glycosylation caused by COG6 gene mutation and literature review
WUBing-bing1,LIWan-xing1,YANGLin,WANGHui-jun,ZHOUWen-hao
(TheMolecularGeneticDiagnosisCenter,ShanghaiKeyLabofBirthDefect,Children'sHospitalofFudanUniversity,Shanghai201102,China; 1Co-firstauthor)
YANG Lin,E-mail:yanglin_fudan@163.com
Objective To diagnose a neonate as congenital disorders of glycosylation caused byCOG6 gene mutation (COG6-CDG),summarize and compare clinical features of 7 subtypes of CDGs caused by mutations of Golgi complex (COG),and to provide the basis for accurate diagnosis,clinical decision-making and outcome prediction of COG-CDGs.MethodsAnalysis was performed on clinical features,parental sanger test,imageological examination,laboratory test and follow-up of a patient carrying a pair of compound heterozygous mutations ofCOG6,and literatures about clinical features of CDGs caused byCOG6 and other COGs were reviewed.ResultsThe patient presented with recurrent hyperpyrexia,elevated liver enzymes,congenital heart disease,prolonged APTT,multiple small lesions in left kidney,micrognathia,no calcification of second molar,eosinophil deficiency.A pair of compound heterozygous mutations ofCOG6 was found by WES.c.511C>T p.R171X was from mother and was reported as a pathogenic mutation ofCOG6-CDG by HGMD,while c.540G>A p.E180E was from father and was a novel splicing mutation.Nine patients were reported asCOG6-CDG listed by HGMD,clinical features were liver dysfunction,abnormal growth and development,facial abnormalities,recurrent hyperpyrexia,hypohidrosis,skin abnormalities,congenital heart disease,renal abnormalities,coagulation abnormalities,abnormal immune system,skeletal joint deformities,seizures,abnormal brain MRI,hearing or visual abnormalities or other malformations,more than half patients died and the rest survivors progressed to severe liver dysfunction with recurrent infections.ConclusionThe first case ofCOG6-CDG is diagnosed and reported in China,COG6-CDG is a rare genetic disease involving multiple organ systems,hypohidrotic ectodermal dysplasia with liver enzyme abnormalities is its main clinical features,most patients present with abnormal growth and development,and poor prognosis.Neonatal gene sequencing can help diagnose COG-CDG and provide the basis for accurate diagnosis,clinical decision-making and outcome prediction of COG-CDGs.
COG6 gene; Congenital disorders of glycosylation; Hypohidrotic ectodermal dysplasia; Mutation; Golgi complex
上海市科学技术委员会基金:15XD1500800
复旦大学附属儿科医院分子诊断中心,上海市出生缺陷防治重点实验室 上海,201102;1 共同第一作者
杨琳,E-mail:yanglin_fudan@163.com
10.3969/j.issn.1673-5501.2017.01.010
2017-01-17
2017-02-13)
