基于生物信息学方法的胰腺导管腺癌预后风险标志物筛选
2017-03-23张志鹏陆晔斌陈泓西夏华孙维佳
张志鹏,陆晔斌,陈泓西,夏华,孙维佳
(中南大学湘雅医院 胆胰外科,湖南 长沙 410008)
胰腺癌是目前最难诊治的恶性肿瘤之一,其病死率几乎与发病率持平。据统计,2015年美国胰腺癌新发48 960例,死亡40 560例,病死率在男女性别中均排第4位[1]。在全球范围内,预计2015年胰腺癌新发病例达到367 000例,死亡359 000例,如果预后得不到改善的话,胰腺癌将在下个10年内成为癌症相关性死亡的第二大主要原因[2-3]。胰腺导管腺癌是胰腺癌中最常见的病理类型,其比例超过90%,而其恶性程度也远高于其他10%,如黏液性囊腺癌和腺泡细胞癌等[4]。由于胰腺导管腺癌早期症状不明显,缺乏特异性的肿瘤标志物,并常伴有局部的神经、血管侵犯及早期远处转移,当确诊时往往已处于进展期,且对放化疗不敏感,导致其总体5年生存率不到5%[5]。只有10%~20%的患者病灶局限,可采取手术治疗,但术后复发率高达80%,其5年生存率也仅为15%~25%[6-7]。虽然近年来在胰腺癌的诊断及治疗上取得了一些进步,但患者的预后并未得到明显的改善[8]。因此,探索胰腺导管腺癌的预后风险标志物可能会为它的治疗提供新的思路。
本研究从癌症基因组数据集(The Cancer Genome Atlas,TCGA)数据库下载了胰腺导管腺癌患者的临床资料、miRNA和gene表达谱数据。通过弹性网络Cox比例风险回归分析(EN-Cox),绘制受试者工作特征(ROC)曲线和Kaplan-Meier曲线,筛选出了与胰腺导管腺癌预后风险明显相关的miRNA和基因;然后对预后风险miRNA的靶基因进行功能预测,对预后风险基因及预后风险miRNA的靶基因进行文献挖掘和功能分析,从而发现潜在的胰腺导管腺癌预后风险标志物。
1 资料与方法
1.1 数据来源
TCGA(https://cancergenome.nih.gov)数据库包含肿瘤患者的临床资料、肿瘤基因组特征和高通量分析数据,而且是迄今世界范围内最大、最成功的癌症基因组数据库。本研究从TCGA数据库中下载了197例胰腺癌患者的临床资料,其中有150例为胰腺导管腺癌。在这150例胰腺导管腺癌中,有2例没有准确的总体生存时间(OS)而被剔除,即有148例具有完整的生存资料。
基于IlluminaHiSeq的高通量测序功能,下载了183例胰腺癌患者的miRNA和基因表达谱数据,包括上述148例胰腺导管腺癌的137例。这137例包含miRNA和基因表达谱数据的胰腺导管腺癌病例用于预后风险标志物的筛选。
1.2 数据预处理
miRNA和基因表达值将以每100万标记读本中每千碱基外显子的读本数(RPKM)[9]进行估算。此外,不同样本间通过中位数法进行标准化处理。
1.3 筛选可能的预后风险miRNA和基因
EN是一种理想的变量选择方法,既能有效处理共线性又可以降维。Cox是一种分析生存资料的半参数模型。EN-Cox则既能处理共线性又可以降维,是分析处理高维小样本生存资料的理想模型。应用EN-Cox筛选回归分析的自变量,构建回归模型,筛选预后风险因素。本研究利用R的glmnet包(https://cran.r-project.org/web/packages/glmnet/index.html)[10]完成EN-Cox回归分析过程,筛选出可能的预后风险miRNA和基因。参数为λ的最小值。
1.4 筛选预后风险标志物
在生物信息学研究中,ROC曲线常被用于评估分层效应,Kaplan-Meier曲线常被用于生存资料的单变量统计分析。在本研究中,利用R(The R Project for Statistical Computing)的pROC包(https://cran.r-project.org/web/packages/pROC/index.html)[11]绘制ROC曲线,确定患者分组的截断值,以此截断值为分界将所有患者分为两组,然后利用R的survival包(https://cran.r-project.org/web/packages/survival/index.html)[12]绘制KM曲线,通过Log-rank检验两组患者的生存时间有无统计学差异。得到有差异的miRNA和基因被认为与胰腺导管腺癌的预后风险明显相关。
1.5 预后风险miRNA的功能预测
在MiRWalk(http://zmf.umm.uni-heidelberg.de/apps/zmf/mirwalk2)[13]数据库中筛选预后风险miRNA的靶基因,并通过Cytoscape(http://www.cytoscape.org)构建miRNA与靶基因的调控网络图。Cytoscape是一种资源开放的软件,可用于数据整合和生物网络的可视化处理[14]。进而,对miRNA的靶基因进行KEGG(Kyoto encyclopedia of genes and genomes)(http://www.kegg.jp)[15]及REACTOME(http://www.reatome.org)[16]信号通路、基因本体论(gene ontology,GO)(http://geneontology.org)[17]和疾病本体论(disease ontology,DO)(http://disease-ontology.org)富集分析。通过TargetMine数据库的超几何分析确定富集的信号通路、生物过程(biological process,BP)、细胞成分(cellular component,CC)、分子功能(molecular function,MF)和疾病名称。TargetMine(http://targetmine.mizuguchilab.org)[18]是一个用于检索靶基因和蛋白的数据库,P<0.05认为具有统计学意义。
1.6 预后风险基因的文献挖掘和功能分析
对预后风险基因及预后风险miRNA的靶基因进行文献挖掘和功能分析。通过TRRUST(http://www.grnpedia.org/trrust)数据库检索预后风险基因的转录活性,并筛选出目标转录因子的靶基因及其协同转录因子,然后用Cytoscape绘制转录调控网络。TRRUST是目前已知最大的且有文献证实的转录调控网络数据库,并可将靶基因的模块化及转录因子的协同性显示出来[19]。
2 结 果
2.1 数据预处理
经过数据预处理后,共得到137例胰腺导管腺癌中的797个miRNA和19 969个基因表达谱数据。
2.2 筛选可能的预后风险miRNA和基因
λ=0.107被设为参数,基于此参数,共筛选出包括5个miRNA和54个基因在内的59个预后风险因素。
2.3 筛选预后风险标志物
根据ROC曲线的截断值,将所有患者分为两组,即miRNA或基因表达值低于截断值者被归于低表达组,miRNA或基因表达值高于截断值者被归于高表达组,共得到16个基因和1个miRNA(表1)。这17个预后风险标志物被认为与胰腺导管腺癌的预后明显相关(均P<0.05)。
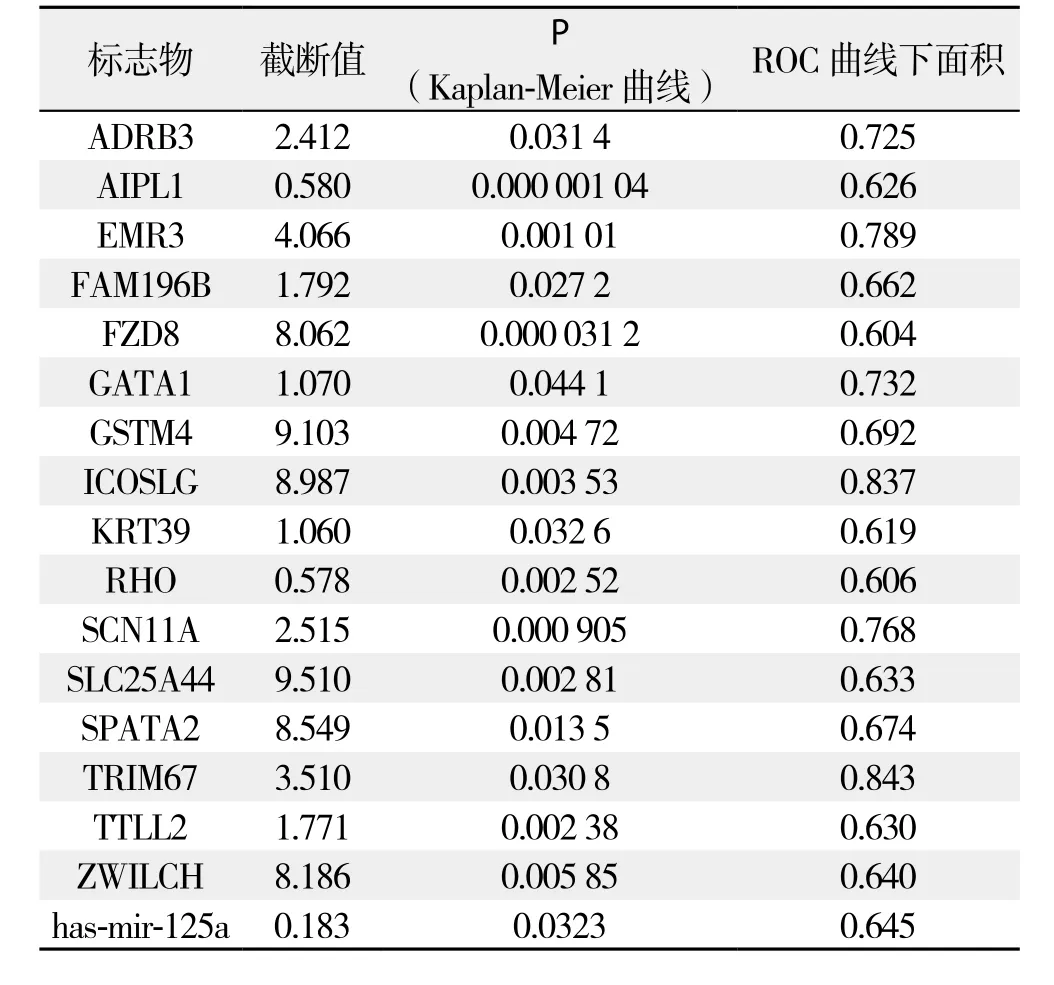
表1 风险标志物筛选结果Table 1 Results of prognostic risk marker identi fi cation
2.4 预后风险miRNA的功能预测
在MiRWalk数据库中检索到miRNA-125a的靶基因有1 982个,其中miRNA-125a-3p有1 030个靶基因,而miRNA-125a-5p有1 021个(图1)。miRNA-125a-3p的靶基因主要富集于甘油脂类代谢通路(KEGG),小分子物质分解代谢过程(BP)和皮肤纤维瘤(DO)中(图2),如成纤维细胞生长因子受体2(fibroblast growth factor receptor 2,FGFR2)富集于皮肤纤维瘤。miRNA-125a-5p的靶基因主要富集于核酸结合区域富亮氨酸重复包含受体通路(KEGG)和淋巴细胞趋化性调控过程(BP)中(图3)。

图1 miRNA-125a及其靶基因的调节网络 (黄色矩形节点代表miRNA-125a-3p和miRNA-125a-5p,粉红色与绿色椭圆节点代表miRNA-125a-3p和miRNA-125a-5p的靶基因,其中绿色椭圆节点同时也是本研究筛选出的预后风险基因)Figure 1 The regulatory network of miRNA-125a and target genes (The yellow rectangle nodes representing miRNA-125a-3p and miRNA-125a-5p, the pink and green ellipse nodes representing the targets genes of miRNA-125a-3p and miRNA-125a-5p, of which the green ellipse nodes also the prognostic risk genes identi fi ed in this study)
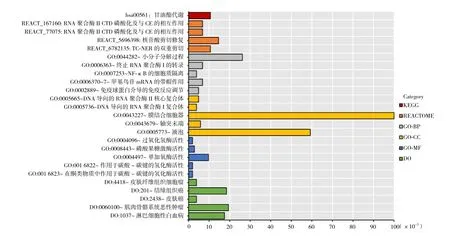
图2 miRNA-125a-3p靶基因的功能富集分析Figure 2 The functional enrichment analyses of the target genes of miRNA-125a-3p

图3 miRNA-125a-5p靶基因的功能富集分析Figure 3 The functional enrichment analyses of the target genes of miRNA-125a-5p
2.5 预后风险基因的文献挖掘和功能分析
miRNA-125a共有1 982个靶基因,其中谷胱甘肽S转移酶μ4(GSTM4)既是miRNA-125a-5p的靶基因又是预后风险基因,可诱导T细胞共刺激分子配体(ICOSLG)、精子发生相关2(SPATA2)既是miRNA-125a-3p的靶基因又是预后风险基因。在16个预后风险基因中,只有GATA结合蛋白1(GATA1)可编码转录因子。基于TRRUST数据库检索到的GATA1的靶基因及协同转录因子,通过Cytoscape软件构建GATA1的转录调控网络(图4)。

图4 GATA1与其靶基因及转录调节因子的转录调节网络(绿色三角节点代表GATA1的靶基因,粉红色矩形节点代表GATA1的转录调节因子)Figure 4 The transcriptional regulatory network of GATA1 and its target genes and transcriptional coregulators (The green triangle nodes representing the target genes of GATA1, and the pink rectangle nodes representing the transcriptional coregulators of GATA1)
3 讨 论
胰腺癌是世界范围内最致命的恶性肿瘤之一,几十年来其治疗效果及预后仍然较差,而其病理类型的90%以上是胰腺导管腺癌[20]。由于胰腺导管腺癌缺乏典型的临床表现及特异性的肿瘤标志物,并常伴有血管神经浸润及早期远处转移和对传统治疗方法的高度抵抗,使得诊断时多处于进展期,严重阻碍了其治疗的实施及预后的改善[21]。因此,探索胰腺导管腺癌的预后风险标志物可能会为它的治疗提供新的思路。
本研究发现了包括1个miRNA和16个基因在内的17个预后风险标志物;同时检索了miRNA的靶基因,并对其靶基因进行功能预测,发现它们主要在皮肤纤维瘤和结缔组织肿瘤中富集。此外,ICOSLG、SPATA2和GSTM4既是预后风险基因又是预后风险miRNA的靶基因。在16个预后风险基因中,只有GATA1可编码转录因子。研究结果提示这些预后风险因子可能参与了胰腺导管腺癌的病理进程。
miRNA-125a是本研究筛选到的唯一的一个miRNA。近年来对miRNA已经进行了大量的研究,其作用方式已经相对明确,已被发现在许多生理及病理过程中发挥了重要作用。miRNA-125a有两种形式:miRNA-125a-3p和miRNA-125a-5p,分别来源于miRNA-125a前体的3'端和5'端。Jiang等[22-23]发现,miRNA-125a的这两种形式均可通过p53信号通路诱导肺癌细胞的凋亡,发挥抑癌基因的作用,不同的是miRNA-125a-3p不完全依靠该信号通路。Hashiguchi等[24]发现miRNA-125a-3p可抑制胃癌细胞的增殖,并与胃癌的临床病理特征呈负相关,可作为一种潜在的预后风险因子。Yang等[25]通过转录组学分析推测miRNA-125a-5p可能参与了胰腺癌的发病机制,并有望成为治疗的新靶点,与本次研究结果一致。
在16个预后风险基因中,ICOSLG、SPATA2和GSTM4又属于miRNA-125a的靶基因,其中ICOSLG、SPATA2为miRNA-125a-3p的靶基因,GSTM4为miRNA-125a-5p的靶基因。ICOSLG,其编码的蛋白质为可诱导T细胞共刺激分子配体(inducible T-cell co-stimulator ligand,ICOSLG),是B7家族成员之一,为新发现的一种细胞表面分子,常表达于B淋巴细胞、树突状细胞及巨噬细胞等抗原递呈细胞上[26]。ICOSLG为ICOS(可诱导T细胞共刺激分子)的唯一配体,后者是CD28超家族的成员,在活化的T细胞及调节性T细胞上呈高表达。生理状态下ICOS通过与细胞表面的ICOSLG相互作用,可促进T细胞的活化、增殖与细胞因子的分泌。近年来的研究[27]显示,ICOS/ICOSLG信号通路参与了炎症反应、自身免疫性疾病及肿瘤的发生。众所周知,肿瘤的发生与免疫逃逸有关。Faget等[28]研究发现ICOS/ICOSLG的相互作用可通过抑制CD4+T淋巴细胞的免疫反应而促进乳腺癌的进展,并且ICOS+细胞的增多与乳腺癌较差的预后相关,从而认为阻断ICOS与ICOSLG的结合可作为乳腺癌临床治疗的新策略。
SPATA2,起初被称为PD1,于1999年在人类睾丸cDNA文库中被首次发现,后来研究发现其表达与精子发生有关,遂更名为SPATA2,其编码的蛋白质为精子发生相关蛋白2[29]。Luca等[30]发现SPATA2可以促进胰岛β细胞的有丝分裂;通过多克隆抗体抑制其表达后,促进有丝分裂的能力明显受限。此外,SPATA2还是NF-κB信号通路中的关键分子,可与关键蛋白CYLD结合,并将其招募至TNF受体复合物;SPATA2的缺失会促进TNF诱导的细胞转录,并抑制其诱导的细胞凋亡,从而导致肿瘤的发生[31]。因此,miRNA-125a-3p及其靶基因ICOSLG、SPATA2在胰腺导管腺癌的作用还有待进一步的阐明。
GSTM4是谷胱甘肽S转移酶(Glutathione S-transferases,GSTs)家族μ亚族的成员,又是miRNA-125a-5p的靶基因。谷胱甘肽S转移酶是一种催化谷胱甘肽与致癌物、药物、毒性物质及氧化应激产物结合,以减少这些物质对细胞成分产生毒性作用的的解毒酶。尤文肉瘤是小儿第二常见的骨与软组织恶性肿瘤,而绝大多数尤文肉瘤是由染色体易位导致的EWS/FLI融合蛋白所引起的[32]。有研究[33]报道GSTM4是EWS/FLI融合蛋白的靶基因,GSTM4表达上调能够促进尤文肉瘤的发生及化疗抵抗,并与不良的预后相关;而GSTM4沉默则可以减少其恶性转化并增强对化疗药物的敏感性,从而认为GSTM4可作为尤文肉瘤的潜在治疗靶点。结合本研究结果,笔者推测miRNA-125a-5p可能通过靶向抑制GSTM4的表达,在胰腺导管腺癌中发挥抑癌作用。
在16个预后风险基因中,只有GATA1可编码转录因子。GATA1,其编码的蛋白质为GATA结合蛋白1(GATA binding protein 1),因能特异性地结合血红蛋白基因调控区域的GATA蛋白而被发现,是GATA家族第一个被发现的成员[34]。GATA1可直接抑制细胞增殖相关基因的表达,如Kit、Myc和Myb等促癌基因,失去对这些靶基因的抑制会导致幼稚红细胞的大量增殖[35]。唐氏综合征(又称21-三体综合征)的患儿有发生急性巨核细胞白血病的高风险。几乎所有发展为急性巨核细胞白血病的唐氏综合征患者都含有GATA1的体细胞突变,而无GATA1突变的唐氏综合征患者则不发展为白血病[36]。因此,GATA1是否突变可作为识别唐氏综合征患儿发生白血病的潜在预测指标,尚未见GATA1在胰腺癌中的研究报道。
除了上述提到的基因外,本研究还发现了其他几个预后风险基因,但在胰腺癌中的作用还有待阐明。总之,本次生物信息学分析将有望为探索胰腺癌的发病机制和治疗提供新的思路和方向,但还需要后续的实验证实。
[1]Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015[J]. CA Cancer J Clin, 2015, 65(1):5–29. doi: 10.3322/caac.21254.
[2]Ferlay J, Soerjomataram I, Ervik M, et al. GLOBOCAN 2012 v1.0,Cancer Incidence and Mortality Worldwide: IARC CancerBase No.11 [Internet]. http://globocan.iarc.fr/Pages/references.aspx
[3]Rahib L, Smith BD, Aizenberg R, et al. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver,and pancreas cancers in the United States[J]. Cancer Res, 2014,74(11):2913–2921. doi: 10.1158/0008–5472.CAN–14–0155.
[4]Yang H, Liu P, Zhang J, et al. Long noncoding RNA MIR31HG exhibits oncogenic property in pancreatic ductal adenocarcinoma and is negatively regulated by miR-193b[J]. Oncogene, 2016,35(28):3647–3657. doi: 10.1038/onc.2015.430.
[5]Onete VG, Besselink MG, Salsbach CM, et al. Impact of centralization of pancreatoduodenectomy on reported radical resections rates in nationwide pathology database[J]. HPB (Oxford),2015, 17(8):736–742. doi: 10.1111/hpb.12425.
[6]Kleeff J, Korc M, Apte M, et al. Pancreatic cancer[J]. Nat Rev Dis Primers, 2016, 2:16022. doi: 10.1038/nrdp.2016.22.
[7]He J, Ahuja N, Makary MA, et al. 2564 resected periampullary adenocarcinomas at a single institution: trends over three decades[J].HPB (Oxford), 2014, 16(1):83–90. doi: 10.1111/hpb.12078.
[8]Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013[J]. CA Cancer J Clin, 2013, 63(1):11–30. doi: 10.3322/caac.21166.
[9]Wagner GP, Kin K, Lynch VJ. Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples[J]. Theory Biosci, 2012, 131(4):281–285. doi: 10.1007/s12064–012–0162–3.
[10]Friedman J, Hastie T, Tibshirani R. Regularization paths for generalized linear models via coordinate descent[J]. J Stat Softw,2010, 33(1):1–22.
[11]Robin X, Turck N, Hainard A, et al. pROC: an open-source package for R and S+ to analyze and compare ROC curves[J]. BMC Bioinforma, 2011, 12(1):77. doi: 10.1186/1471–2105–12–77.
[12]Therneau TM. A Package for Survival Analysis in S. R package version 2.37–7, URL http://CRAN.R-project.org/package=survival.
[13]Dweep H, Sticht C, Pandey P, et al. miRWalk–database: prediction of possible miRNA binding sites by “walking” the genes of three genomes[J]. J Biomed Inform, 2011, 44(5):839–847. doi: 10.1016/j.jbi.2011.05.002.
[14]Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks[J]. Genome Res, 2003, 13(11):2498–2504.
[15]Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes[J]. Nucleic Acids Res, 2000, 28(1):27–30.
[16]Croft D, O’Kelly G,Wu G, et al. Reactome: a database of reactions, pathways and biological processes[J]. Nucleic Acids Res,2011, 39(Database issue):D691–697. doi: 10.1093/nar/gkq1018.
[17]Ashburner M, Ball CA, Blake JA, et al. Gene ontology: tool for the uni fi cation of biology. The Gene Ontology Consortium[J]. Nat Genet, 2000, 25(1):25–29.
[18]Chen YA, Tripathi LP, Mizuguchi K. TargetMine, an integrated data warehouse for candidate gene prioritisation and target discovery[J].PLoS One, 2011, 6(3):e17844. doi: 10.1371/journal.pone.0017844.
[19]Han H, Shim H, Shin D, et al. TRRUST: a reference database of human transcriptional regulatory interactions[J]. Sci Rep, 2015,5:11432. doi: 10.1038/srep11432.
[20]Yonemori K, Kurahara H, Maemura K, et al. MicroRNA in pancreatic cancer[J]. J Hum Genet, 2017, 62(1):33–40. doi:10.1038/jhg.2016.59.
[21][No authors listed]. Pancreatic cancer[J]. Nat Rev Dis Primers,2016, 2:16023. doi: 10.1038/nrdp.2016.23.
[22]Jiang L, Huang Q, Chang J, et al. MicroRNA HSA-miR-125a-5p induces apoptosis by activating p53 in lung cancer cells[J].Exp Lung Res, 2011, 37(7):387–398. doi: 10.3109/01902148.2010.492068.
[23]Jiang L, Chang J, Zhang Q, et al. MicroRNA hsa-miR-125a-3p activates p53 and induces apoptosis in lung cancer cells[J]. Cancer Invest, 2013, 31(8):538–544. doi: 10.3109/07357907.2013.820314.
[24]Hashiguchi Y, Nishida N, Mimori K, et al. Down-regulation of miR-125a-3p in human gastric cancer and its clinicopathological signi fi cance[J]. Int J Oncol, 2012, 40(5):1477–1482. doi: 10.3892/ijo.2012.1363.
[25]Yang J, Zeng Y. Identification of miRNA-mRNA crosstalk in pancreatic cancer by integrating transcriptome analysis[J]. Eur Rev Med Pharmacol Sci, 2015, 19(5):825–834.
[26]He M, Wang Y, Shi WJ, et al. Immunomodulation of inducible costimulator (ICOS) in human cytokine-induced killer cells against cholangiocarcinoma through ICOS/ICOS ligand interaction[J]. J Dig Dis, 2011, 12(5):393–400. doi: 10.1111/j.1751–2980.2011.00527.x.
[27]Merrill JT. Co-stimulatory molecules as targets for treatment of lupus[J]. Clin Immunol, 2013, 148(3):369–375. doi: 10.1016/j.clim.2013.04.012.
[28]Faget J, Bendriss-Vermare N, Gobert M, et al. ICOS-ligand expression on plasmacytoid dendritic cells supports breast cancer progression by promoting the accumulation of immunosuppressive CD4+ T cells[J]. Cancer Res, 2012, 72(23):6130–6141. doi:10.1158/0008–5472.CAN–12–2409.
[29]Maran C, Tassone E, Masola V, et al. The Story of SPATA2(Spermatogenesis-Associated Protein 2): From Sertoli Cells to Pancreatic Beta-Cells[J]. Curr Genomics, 2009, 10(5):361–363. doi:10.2174/138920209788920976.
[30]Luca G, Calvitti M, Baroni T, et al. Sertoli cell-induced adult rat islet beta-cell mitogenesis: causative pathways[J]. Diabetes Nutr Metab, 2003, 16(1):1–6.
[31]Schlicher L, Wissler M, Preiss F, et al. SPATA2 promotes CYLD activity and regulates TNF-induced NF-κB signaling and cell death[J]. EMBO Rep, 2016, 17(10):1485–1497.
[32]Toomey EC, Schiffman JD, Lessnick SL. Recent advances in the molecular pathogenesis of Ewing's sarcoma[J]. Oncogene, 2010,29(32):4504–4516. doi: 10.1038/onc.2010.205.
[33]Luo W, Gangwal K, Sankar S, et al. GSTM4 is a microsatellitecontaining EWS/FLI target involved in Ewing's sarcoma oncogenesis and therapeutic resistance[J]. Oncogene, 2009,28(46):4126–4132. doi: 10.1038/onc.2009.262.
[34]Evans T, Felsenfeld G. The erythroid-speci fi c transcription factor Eryf1: a new fi nger protein[J]. Cell, 1989, 58(5):877–885.
[35]Munugalavadla V, Dore LC, Tan BL, et al. Repression of c-kit and its downstream substrates by GATA-1 inhibits cell proliferation during erythroid maturation[J]. Mol Cell Biol, 2005, 25(15):6747–6759.
[36]Roberts I, Alford K, Hall G, et al. GATA1-mutant clones are frequent and often unsuspected in babies with Down syndrome:identi fi cation of a population at risk of leukemia[J]. Blood, 2013,122(24):3908–3917. doi: 10.1182/blood–2013–07–515148.
