过渡金属催化脱羰反应研究进展
2017-03-22严绍熙
严绍熙
过渡金属催化脱羰反应研究进展
严绍熙
(韶关学院化学与环境工程学院, 广东 韶关 521005)
羰基是有机化合物中重要的官能团,对于这类化合物,一般都需要在过渡金属的催化下才能使它们脱去羰基。文章中我们将对近年来部分脱羰反应进行介绍。
过渡金属; 脱羰; 偶联反应
Heck反应和Suzuki交叉偶联反应被科研工作者广泛应用于有机合成中,为了避免反应类型的限制,必须进一步拓展偶联反应,为此,过渡金属催化的脱羰偶联反应吸引了研究者的兴趣。本文通过对近年来金属催化下的脱羰偶联反应进行简要的综述。
1 金属催化的酰卤类的脱羰偶联反应
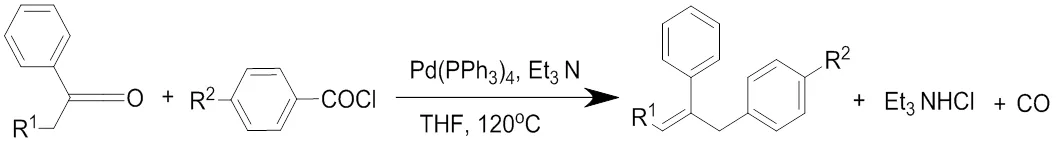
芳基酰卤化合物在有机反应中是比较活泼的,但是在脱羰反应中依然需要在贵价的过渡金属催下才能反应。在1987年酰氯的脱羰反应首次被发现的,Watanabe等[1]在合成1,2-不饱和醛酮时发现:在四三苯基磷钯作催化剂、三乙胺作碱、四氢呋喃作为溶剂体系催化下,酰氯化合物发生脱羧反应,与苯醌化合物偶联,形成目标化合物,同时形成副产物氯化铵和一氧化碳。
随着金属催化反应的发展,酰氯化合物脱羧反应得到了一定的重视:Andrus[2-3]等利用酰氯在醋酸钯、配体、碱的作用下发生脱羰偶联反应合成具有抗癌作用的天然药物分子白藜芦醇,这也是脱羰反应首次应用在天然药物分子合成中。
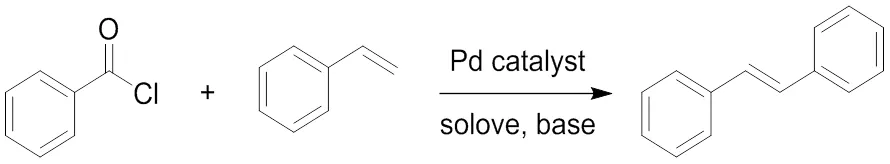
金属催化下得到的乙烯类化合物,通常是利用芳基卤和芳基乙烯在金属钯的催化下发生Heck反应形成的,但是该类型反应对于富电子的芳基乙烯反应不佳。2007年,Martins等[4]在微波条件下利用苯酰氯脱羧与苯乙烯偶联合成二苯乙烯。
2008年,Yu等[5]首次报道了苯并喹啉特定位置上的氢被活化与芳基酰氯发生脱羧偶联反应。这个反应是在[Rh(COD)Cl]2作为催化剂、碳酸钠作碱、二甲苯作反应溶剂的体系下发生的,同时该反应必须添加4A分子筛,否则不能够得到目标产物,可见分子筛的重要性;另外反应温度高达145oC,不利于操作。作者提出的可能机理,认为芳基酰氯2与一价的铑发生氧化加成形成化合物4,接着在高温下发生脱羧反应形成中间体5,然后在碱的作用下苯并喹啉1得到活化与5形成分子内中间体6,接着发生还原反应形成目标产物3。2010年,作者把该反应拓展到芳基乙烯酮和酰氯的脱羰偶联反应[7],为合成乙烯类化合物增添多一种途径。

2010年Ellman等[7]把此类特定位置活化的反应应用到吡啶类化合物。这为以后杂环类化合物的合成寻找了新的研究方向,不过存在收率不高的缺点。
2 金属催化的脂类脱羰反应
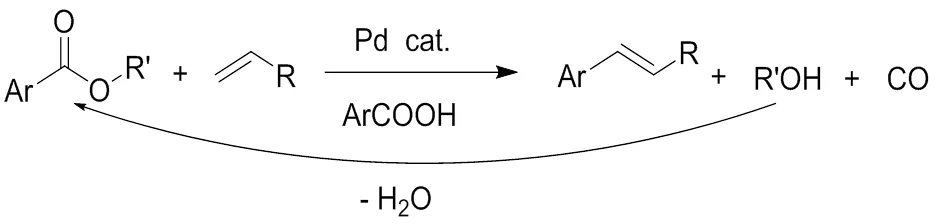
据研究,羧酸的脱羧反应在比较廉价的过渡金属催下就可以发生。而酯类相对稳定,要发生该类反应都要在贵价金属的催化下。2002年Lukas等[8]首次报道脂类的脱羰偶联反应,该反应在钯催化下发生的,并且副产物只有一氧化碳和水,而另一产物醇则可以重复利用。
2012年,Amaike等[9]报道了通过使用廉价镍作为催化剂使到芳基脂脱羧,这一反应没有使用双金属催化,不过反应温度高并且使用了大量的配体和碱。2015年,Muto1等[10]对该反应进一步拓展,有效的把底物拓展到杂环类芳基脂。
3 金属催化的醛类的脱羰偶联反应
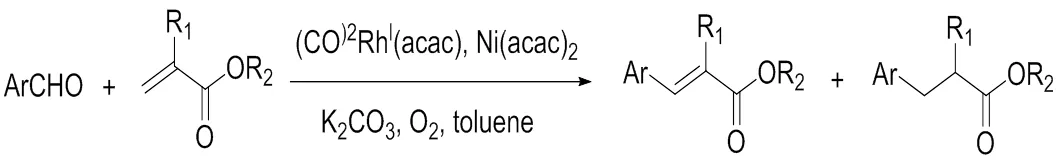
直到2010年,醛类的脱羰偶联反应首次被报道,此类反应主要是由李超军课题组研究的[11-15],所使用的催化金属涉及到铱、铷、铑、镍、铜等金属。在这类反应中可以发现大部分反应都需要在双金属催化、配体、添加剂以及高温的条件下才发生,说明醛基的脱羧反应条件比较苛刻。
4 结束语
随着金属催化反应的发展,过渡金属催化的脱羰偶联反应渐渐的被科研人员深入研究,特别是天然药物分子白藜芦醇的合成。虽然已经报道了多种的脱羧反应的方法,但这些方法存在着一定的缺陷,例如:使用贵金属催化、反应温度较高、反应时间久、使用大量的碱,而且有些反应条件比较苛刻,不利于推广。因此探索廉价金属催化的脱羧反应具有重要的意义。
[1]Mitsudo, T.; Kadokura, M.; Watanabe Y. Novel Synthesis of α,β-Unsaturated Ketones by the Palladium-Catalyzed Arylation of Ketenes with Aroyl Chlorides or the Decarbonylative Cross-Condensation of Acyl Halides [J]. J. Org. Chem. 1987, 52: 3186-3192.
[2]Andrus, M. B.; Liu, J.; Meredith, E. L.; et al. Synthesis of resveratrol using a direct decarbonylative Heck approach from resorcylic acid [J]. Tetrahedron Lett. 2003, 44: 4819-4822.
[3]Andrus, M. B.; Liu, J. Synthesis of polyhydroxylated ester analogs of the stilbene resveratrol using decarbonylative Heck couplings [J]. Tetrahedron Lett. 2006, 47: 5811-5814.
[4]Martins, D. L.; Alvarezb, H. M.; Aguiara, C.S.; et al. Palladium Catalyzed Decarbonylative Mizoroki-Heck Reactions of Benzoyl Chloride and Styrene Under Microwave Irradiation [J]. Lett Org Chem, 2007, 4: 253-258.
[5]Zhao, X. D.; Yu, Z.K. Rhodium-Catalyzed Regioselective C-H Functionalization via Decarbonylation of Acid Chlorides and C-H Bond Activation under Phosphine-Free Conditions [J]. J. Am. Chem. Soc. 2008, 130: 8136-8137.
[6]Ye, W. J.; L, N.; Yu, Z. K. Rhodium-Catalyzed Direct Alkenylation and Arylation of Arene C-H Bonds via Decarbonylation of Cinnamoyl Chlorides,Cinnamic Anhydrides, and Poly(aroyl) Chlorides [J]. Organometallics. 2010, 29: 1051-1054.
[7]Berman, A. M.; Bergman, R. G.; Ellman. J.A. Rh(I)-Catalyzed Direct Arylation of Azines [J]. J. Org. Chem. 2010, 75: 7863-7868.
[8]Gooben, L. J.; Paetzold, J. Pd-Catalyzed Decarbonylative Olefination of Aryl Esters: Towards a Waste-Free Heck Reaction [J]. Angew. Chem. Int. Ed. 2002, 41: 1237-1241.
[9]Amaike, K.; Muto, K.; Yamaguchi, J.; et al. Decarbonylative C-H Coupling of Azoles and Aryl Esters: Unprecedented Nickel Catalysis and Application to the Synthesis of Muscoride A [J]. J. Am. Chem. Soc. 2012, 134, 13573-13576
[10]Muto1, K.; Yamaguchi1, J.; Musaev, D. G.; et al. Decarbonylative organoboron cross-coupling of esters by nickel catalysis. Nature Communications, 2015, 7508: 1-8.
[11]Yang, L.; Correia, C.A.; Guo, X.Y.; et al. A novel catalytic decarbonylative Heck-type reaction and conjugate addition of aldehydes to unsaturated carbonyl compounds [J]. Tetrahedron Lett. 2010, 51: 5486-5489.
[12]Guo, X. Y.; Wang, J.; Li, C. J. Ru-Catalyzed Decarbonylative Addition of Aliphatic Aldehydes to Terminal Alkynes [J]. Org. Lett. 2010, 12: 3176-3178.
[13]Guo, X. Y.; Wang, J.; Li, C. J. An Olefination via Ruthenium-Catalyzed Decarbonylative Addition of Aldehydes to Terminal Alkynes [J]. J. Am. Chem. Soc 2009, 131: 15092-15093.
[14]Shuai, Q.; Yang, L.; Guo, X. Y.; et al. Rhodium-Catalyzed Oxidative C-H Arylation of 2-Arylpyridine Derivatives via Decarbonylation of Aromatic Aldehydes [J]. J. Am. Chem. Soc. 2010, 132: 12212-12213.
[15]Guo, X. Y.; Wang, J.; Li, C. J. Iridium as a general catalyst for the decarbonylative addition of aldehydes to alkynes [J]. J. Org. Chem. 2011, 696: 211-215.
Research Progress in Transition Metal-catalyzed Decarbonylative Reactions
(Shaoguan University, Guangdong Shaoguan 512005, China)
The carbanyl group is an important functional group of organic compounds. The transition metal is generally uses as catalyst to remove the carbanyl group in carbonyl compounds. In this paper, some decarbonylation reactions in recent years were summarized.
transition metal; decarbonylation; coupling reaction
TQ 032
A
1004-0935(2017)10-1030-03
本研究得到2015年度韶关学院校级科研项目经费资助。
2017-08-03
严绍熙(出生年1986-),男,民族,助教,硕士学位,研究方向:有机化学。
