反相高效液相色谱法分离分析奥美拉唑及其相关组分
2017-03-19刘静关瑾石爽阎峰
刘静,关瑾,石爽,阎峰
反相高效液相色谱法分离分析奥美拉唑及其相关组分
刘静,关瑾,石爽,阎峰
(沈阳化工大学 应用化学学院,辽宁 沈阳 110142)
建立奥美拉唑及其两种相关组分的反相高效液相色谱分离分析方法。考察了缓冲盐的种类、浓度和pH等色谱条件对分离分析的影响。以乙腈-15 mmol﹒L-1磷酸二氢钠(pH 7.0)为流动相,在C18色谱柱上,采用梯度洗脱,对奥美拉唑及两种相关组分进行分离分析。在优化色谱条件下,奥美拉唑及两种相关组分在10 min内达到基线分离;奥美拉唑、2-巯基-5甲氧基-1H-苯并咪唑(杂质A)和奥美拉唑硫醚(杂质B)在各自测定的质量浓度范围内与峰面积的线性关系良好(2≥0.9960), 检出限(信噪比为3)分别为15 ng﹒mL-1、15 ng﹒mL-1和30 ng﹒mL-1;样品的回收率为98.4%~102.0%,相对标准偏差为0.62%~0.82%。该方法操作简单、快速、准确可靠,并用于实际样品的分析,获得了令人满意的结果。
奥美拉唑;反相高效液相色谱法;分离分析
奥美拉唑为世界上第一个质子泵抑制剂药物,用于治疗消化性溃疡[1-2]。该药能选择性地抑制胃壁细胞的 H+/K+-ATP酶,从而抑制胃酸的分泌,具有疗效高、耐受性好等优点[3-4]。2-巯基-5-甲氧基-1H-苯并咪唑(杂质A)和奥美拉唑硫醚(杂质B)为奥美拉唑的合成原料和中间体[5]。目前文献报道的奥美拉唑的分析方法主要有紫外分光光度计法[6]、高效液相色谱法[7-10]、近红外光谱法[11]和气相色谱法[12]。本文采用反相高效液相色谱法同时分离分析奥美拉唑、杂质A和杂质B,并进行了方法学验证。该方法操作简单、快速、准确可靠,可用于奥美拉唑的质量控制。
1 仪器与材料
Agilent 1260高效液相色谱仪(包括DAD检测、化学工作站,美国安捷伦公司)。奥美拉唑(含量质量分数0.62%≥98.0%)、2-巯基-5甲氧基-1H-苯并咪唑(含量质量分数≥99.0%),奥美拉唑硫醚(含量质量分数≥99.0%)对照品(HPLC面积归一化法测定其含量质量分数),奥美拉唑原料药由沈阳化工大学精细化工实验室制备;乙腈(色谱纯)和其他试剂(分析纯)(天津市大茂化学试剂厂);水为二次重蒸水。
2 方法与结果
2.1 色谱条件
色谱柱:Hypersil ODS2-C18柱(150 mm×4.6 mm,5μm);流动相:乙腈(A)-15 mmol﹒L-1磷酸二氢钠(B)(pH 7.0);梯度洗脱程序: 0~7min: 35% A~60% A,7~10 min: 60% A~35% A,10~15 min: 35% A;流速:1 mL﹒min-1;检测波长: 302 nm;柱温:30 ℃;进样量:20 μL。
2.2 溶液的配制
2.2.1 对照品储备溶液的配制
精密称取奥美拉唑、杂质A、杂质B对照品各20 mg置10 mL容量瓶中,用甲醇将其溶解、定容、摇匀,制得浓度为2 mg﹒mL-1的标准储备液。
2.2.2 供试品溶液的配制
精密量取奥美拉唑、杂质A和杂质B标准储备液适量,制得奥美拉唑和杂质B浓度均为50 μg﹒mL-1、杂质A浓度为15 μg﹒mL-1的混合供试品溶液。
2.3 色谱条件的优化与系统适用性试验
实验考察了乙腈、甲醇对奥美拉唑、杂质A和杂质B分离分析的影响,乙腈表现出最佳分离效果。由于3个组分的极性相差较大,采用等度洗脱程序分离分析时间长,本实验采取梯度洗脱程序。
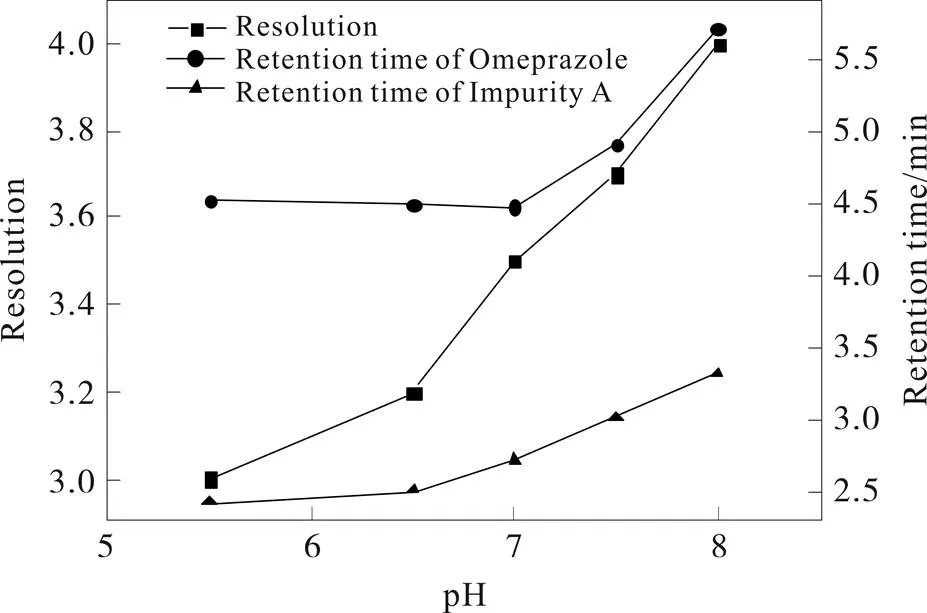
图1 缓冲溶液pH对奥美拉唑和杂质A保留时间和分离度的影响
实验考察了流动相中乙酸盐和磷酸盐溶液对分离分析的影响,磷酸盐体的分离系更稳定。实验进一步考察了磷酸盐浓度和pH对分离分析的影响,比较磷酸盐浓度为10、15、20、25、30 mmol﹒L-1时对分离分析的影响,磷酸盐浓度为15 mmol﹒L-1时,分离效果最好。实验考察磷酸盐溶液的pH对分离的影响,由图1可见,随着pH增大,分离度增大,但是保留时间延长。综合考虑分离度和分析时间,选择pH为7。
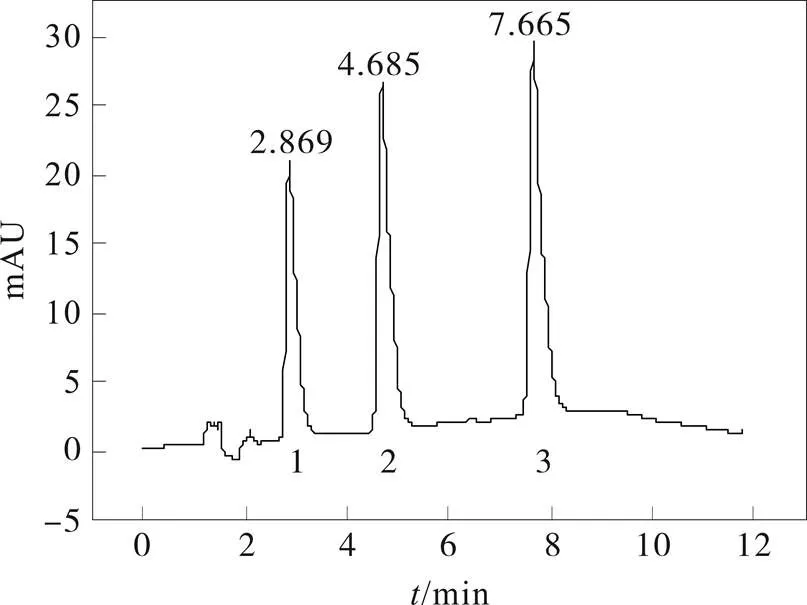
图2 奥美拉唑及杂质A、杂质B的HPLC色谱图
1- 杂质A;2- 奥美拉唑;3-杂质B
以乙腈-15 mmol﹒L-1磷酸二氢钠(pH 7.0)为流动相,在C18色谱柱上,采用梯度洗脱,奥美拉唑、杂质A和杂质B的分离色谱图见图2,各色谱峰分离度均大于3,理论塔板数不小于5 000。
2.4 方法学考察
2.4.1 方法的线性范围、检测限和定量限
配制奥美拉唑系列标准溶液,浓度范围为1 ~ 1 200 μg﹒mL-1;杂质A系列标准溶液,浓度范围为0.05~1 μg﹒mL-1;杂质B系列标准溶液,浓度范围为0.12~2.4 μg﹒mL-1。以目标分析物峰面积为纵坐标()、质量浓度为横坐标(,μg﹒mL-1)绘制标准曲线,其线性范围、回归方程和相关系数(2) 见表1。2均大于0.996 0,表明奥美拉唑、杂质A和杂质B在各自的线性范围内呈良好的线性关系。分别以3倍和10倍信噪比(S/N)确定方法的检测限和定量限,奥美拉唑、杂质A和杂质B的检测限分别为15、15、30 ng﹒mL-1,定量限分别为30、50、120 ng﹒mL-1。

表1 奥美拉唑、杂质A和杂质B的线性范围、线性方程、相关系数
2.4.2 精密度试验
制备800、1 000、1 200 μg﹒mL-1的奥美拉唑溶液各6份;0.05、1 μg﹒mL-1的杂质A溶液各6份;0.12、1 μg﹒mL-1的杂质B溶液各6份,按“2.1”节色谱条件测定峰面积和保留时间,奥美拉唑、杂质A、杂质B峰面积和保留时间的日内精密度的RSD分别均小于2.79%、1.65%,连续分析3天,峰面积和保留时间的日间精密度的RSD分别均小于2.40%、0.93%,表明仪器精密度良好。
2.4.3 回收率试验
制备浓度为50 μg·mL-1的奥美拉唑样品溶液9份,分别添加浓度为25、500和1 000 μg﹒mL-1的标准样品溶液各3份,测定回收率。在1 mg﹒mL-1奥美拉唑样品溶液中按0.1%的杂质限度(杂质的质量浓度为1 μg﹒mL-1)加入杂质A和杂质B,每个样品制备6份,测定杂质A和杂质B的回收率。本方法测得回收率结果为98.4%~102.0%,RSD为0.62%~0.82%,此方法准确度良好。
2.4.4 真实样品含量测定
准确称取实验室自制奥美拉唑样品10 mg,置于10 mL容量瓶中,用甲醇定容,按照“2.1”节的色谱条件进行含量测定,共测试 3批,结果见表2。

表2 真实样品含量测定结果
奥美拉唑含量在97.5%~99.3%,杂质A和杂质B含量在定量限以下。
3 结论
本文建立了奥美拉唑及两种相关组分分离分析的反相高效液相色谱法。以乙腈-15 mmol﹒L-1磷酸二氢钠(pH 7.0)为流动相,采用梯度洗脱对奥美拉唑、杂质A和杂质B进行了分离分析,奥美拉唑、杂质A和杂质B在10 min内达到基线分离。该方法具有操作简便、快速等优点,可用于奥美拉唑的质量控制。
[1]陈玉萍. 奥美拉唑在消化性溃疡并上消化道出血治疗中的应用[J]. 实用预防医学, 2007, 14 (5): 1520-1521.
[2]DEMERTZIS K, POLYMEROS D, EMMANUEL T, et al.Omeprazole maintenance therapy prevents recurrent ulcer bleeding after surgery for duodenal ulcer [J]. World J Gastroenterol,2006,12(5):791-795.
[3]贾斌. 奥美拉唑的药学药理及临床应用效果评价[J]. 中国冶金工业医学杂志, 2016, 33 (4): 488-489.
[4]刘金鹏.奥美拉唑的药理和临床应用探析[J]. 中国现代药物应用, 2016, 10 (6): 130-131.
[5]颜国和, 王飞武. 奥美拉唑合成路线图解[J]. 中国医药工业杂志, 1991, 22 (6): 283-284.
[6]陈新玲,许晓,翟卫芳.紫外-可见分光光度法测定注射用奥美拉唑钠中间品中奥美拉唑的含量[J].首都医药,2009,9:52.
[7]陶巧凤,陈雪帆.奥美拉唑原料及其肠溶胶囊有关物质检查方法的研究[J].药物分析杂志,2005,25(5):576-578.
[8]GALLINELLA B, FERRETTI R, ZANITTI L, et al.Comparison of reversed-phaseenantio selective HPLC methods for determining the enantiomeric purity of (S)-omeprazole in the presence of its related substances[J]. Journal of Pharmaceutical Analysis,2016,6:132-136.
[9]马延,王媛,罗智敏,等.HPLC法测定埃索美拉唑钠的有关物质[J].西北药学杂质,2016,31(2):154-157.
[10]MOHAMED I,WALASH, FAWZIA I, et al .Isocratic RP-HPLC method for separation and simultaneous determination of ternary mixture of omeprazole, tinidazole and doxycycline in their raw materials and combined capsules[J]. The Royal Society of Chemistry, 2013, 5: 5105-5111.
[11]林翔,汪学楷,代涛,等.近红外光谱法应用于奥美拉唑含量分析[J].化学研究与应用,2011,23(7):898-901.
[12]姜鹰雁, 耿志鹏, 刘燕, 等. 顶空气相色谱法同时测定艾司奥美拉唑镁原料药中6种有机溶剂的残留量[J]. 中国药房, 2017, 28 (15): 2093-2096.
Separation and Analysis of Omeprazole and Its Related Components by Reversed-Phase High Performance Liquid Chromatography
,,,
(Shenyang University of Chemical Technology, Liaoning Shenyang 110142, China)
A method for separation and analysis of omeprazole and its two related components by high performance liquid chromatography was established. The effect of buffer salt type and concentration,pH and other chromatographic conditions on the separation and analysis was investigated.Acetonitrile-15 mmol·mL-1phosphate(pH 7) was used as mobile phase,the omeprazole and two related components was separated and analyzed on the C18 column by gradient elution. Under optimized chromatographic conditions,omeprazole and two related components achieved baseline separation within 10 min;In the range of a certain mass concentration,omeprazole, 2-mercapto-5-methoxy-1H-benzimidazole(impurity A)and omeprazole thioether(impurity B) showed good linearity with their peak area respectively(2≥0.9960),their detection limits (signal-to-noise ratio was 3) were15 ng·mL-1, 15 ng·mL-1and 30 ng·mL-1,respectively;the recovery of the sample was 98.4%~102.0%,and the relative standard deviation was 0.62%~0.82%. The method is simple,fast,accurate and reliable, so it can be applied to analyze real samplesand satisfactory results can be obtained.
omeprazole; reversed-phase high performance liquid chromatography; separation and analysis
2017-10-11
刘静(1991-),女,硕士研究生,内蒙古赤峰人,2018年毕业于沈阳化工大学化学专业,研究方向:应用分析。
关瑾(1967-),女,锡伯族,副教授,博士,研究方向:应用化学。
辽宁省自然科学基金,项目号:2015020701。
R917
A
1004-0935(2017)12-1222-03
