高效液相色谱-四极杆/线性离子阱质谱测定替米沙坦中N-甲基邻苯二胺残留量
2017-03-13周长朋王东武郑文凤梁晓艳
周长朋,王东武,许 洁,郑文凤,梁晓艳
(迪沙药业集团国家认定企业技术中心,山东 威海 264205)
高效液相色谱-四极杆/线性离子阱质谱测定替米沙坦中N-甲基邻苯二胺残留量
周长朋*,王东武,许 洁,郑文凤,梁晓艳
(迪沙药业集团国家认定企业技术中心,山东 威海 264205)
建立了高效液相色谱-四极杆/线性离子阱质谱(HPLC-QTRAP-MS/MS)测定替米沙坦中N-甲基邻苯二胺残留量的分析方法。替米沙坦药物以水-乙腈(80∶20,体积比)溶解后,采用Agilent Eclipse XDB-C18色谱柱(3.5 μm,2.1 mm×100 mm)进行分离,以0.1%(体积分数)甲酸水和乙腈作为流动相进行梯度洗脱,电喷雾正离子(ESI+)扫描方式下选择离子监测(SRM)模式对样品进行检测。结果表明,N-甲基邻苯二胺在2.0~50 μg/L 范围内线性关系良好(r>0.99),检出限(S/N=3)为0.5 μg/L ,定量下限(S/N=10)为2.0 μg/L。该方法操作简单、灵敏度高、重现性好,可用于替米沙坦中N-甲基邻苯二胺残留量的测定。
高效液相色谱-四极杆/线性离子阱质谱仪;N-甲基邻苯二胺;替米沙坦
替米沙坦(Telmisartan)是一种特异性血管紧张素Ⅱ受体(AT1型)拮抗剂,其化学名为4-{[2-正丙基-4-甲基-6-(1-甲基苯并咪唑-2-基)苯并咪唑-1-基]甲基}联苯基-2-羧酸(合成路线见图1),具有激动过氧化物酶体增殖物激活受体γ(PPAR-γ)及显著改善血管内皮功能的作用,且能抑制血管平滑肌(VSMC)收缩和增殖,是目前较为常用的抗高血压药物[1-3]。
N-甲基邻苯二胺是替米沙坦合成的起始物料,具有1个苯胺结构,且有一定的致癌毒性,依照欧洲基因毒性杂质的指导原则其应属于潜在基因毒性杂质[4],必须进行严密的控制。参照欧洲EMEA提出的采用“毒理学关注阈值”(Threshold of toxicological concern,TTC)作为基因毒性杂质可接受限度的计算方法[5],N-甲基邻苯二胺的限度为5.0 mg/L,若以替米沙坦的配制浓度为1.0 mg/mL计算,则实际检测浓度应为5.0 μg/L,对检测水平的要求较高。目前针对邻苯二胺的检测方法主要有气相色谱法[6]、液相色谱法[7]、荧光光谱滴定法[8]、化学发光法[9]等,上述方法的检出限均较高。而对于药物中基因毒杂质N-甲基邻苯二胺的研究相对较少,其中王金朝等[10]利用液相色谱法检测了替米沙坦中的N-甲基邻苯二胺残留,但其检出限为40 ng/mL,难以满足当前的限度要求。而国外已有多篇文献报道利用液相色谱-质谱联用仪检测药物中基因毒杂质[11-12],国内也有利用气质联用仪[13-14]、液质联用仪[15-16]等监控基因毒性杂质的方法报道,但对于N-甲基邻苯二胺的液质联用检测方法尚未见报道。
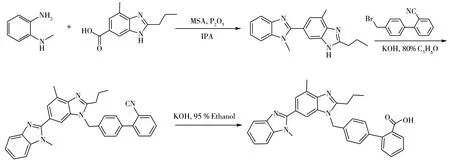
图1 替米沙坦的合成路线图Fig.1 Graphical synthetic routes of telmisartan
四极杆-线性离子阱(QTRAP-MS/MS)质谱仪是一种混合型串联质谱,具有与传统三重四极杆质谱仪相当的定量功能,且灵敏度和特异性均较高,现已广泛应用于食品、药品、环境等领域[17-22]。本文基于高效液相色谱-四极杆/线性离子阱质谱(HPLC-QTRAP-MS/MS)技术,建立了测定替米沙坦药物中基因毒性杂质N-甲基邻苯二胺的分析方法。该方法操作简单、回收率好、灵敏度高,填补了国内替米沙坦药物N-甲基邻苯二胺定量检测研究的空白,有利于对替米沙坦药物合成过程中的质量参数进行有效监测。
1 实验部分
1.1 仪器与试剂
Thermo LTQ-XL型高效液相色谱-四极杆/离子阱质谱仪:配有电喷雾离子源;超声波清洗机(昆山超声仪器公司);BT-25S精密分析天平(瑞士梅特勒-托利多公司 );Milli-Q超纯水仪(美国Millipore公司);0.22 μm有机系滤膜(天津市津腾实验设备有限公司)。
N-甲基邻苯二胺盐酸盐(纯度≥99%,购自Sigma-Aldrich公司);乙腈(色谱纯,德国Merck公司);甲酸(优级纯,天津大茂化学试剂厂);实验用水为Milli-Q超纯水(电阻率为18.2 MΩ·m);替米沙坦精品(迪沙药业集团有限公司)。
1.2 溶液的制备
对照品溶液:取N-甲基邻苯二胺盐酸盐适量,精密称定,至10 mL容量瓶中,加少量乙腈超声溶解,用乙腈定容至刻度,配制成含N-甲基邻苯二胺1.0 g/L的标准储备液,于-18 ℃避光保存。取适量标准储备液,用乙腈配制成10.0 mg/L的标准中间液,作为对照品溶液,于4 ℃避光保存。上机前用水-乙腈(体积比80∶20)稀释成相应浓度,过0.22 μm滤膜后注入质谱。
供试品溶液:取替米沙坦精品约10 mg,精密称定,至10 mL容量瓶中,加少量乙腈超声溶解后,再加入水-乙腈(80∶20)定容至刻度,过0.22 μm滤膜后注入质谱。
1.3 色谱条件
Agilent Eclipse XDB-C18色谱柱(3.5 μm,2.1 mm×100 mm);流动相:A为0.1%(体积分数)甲酸水溶液,B为乙腈;流速:0.2 mL/min;梯度洗脱程序:0~ 2.00 min,20% B;2.00~4.00 min,20%~95% B;4.00~9.00 min,95% B;9.01~15.00 min,20% B;柱温35 ℃;进样体积为10 μL。
1.4 质谱条件
离子源:电喷雾离子源(ESI);扫描方式:正离子扫描;检测方式:选择离子检测(SRM);离子源温度:320 ℃;辅助气温度:300 ℃;鞘气:35 L/min;辅助气:15 L/min;毛细管电压:3 500 V;定量方式:外标法定量;定量离子对:m/z123.1/107.6,碰撞能量(CE):35 V。
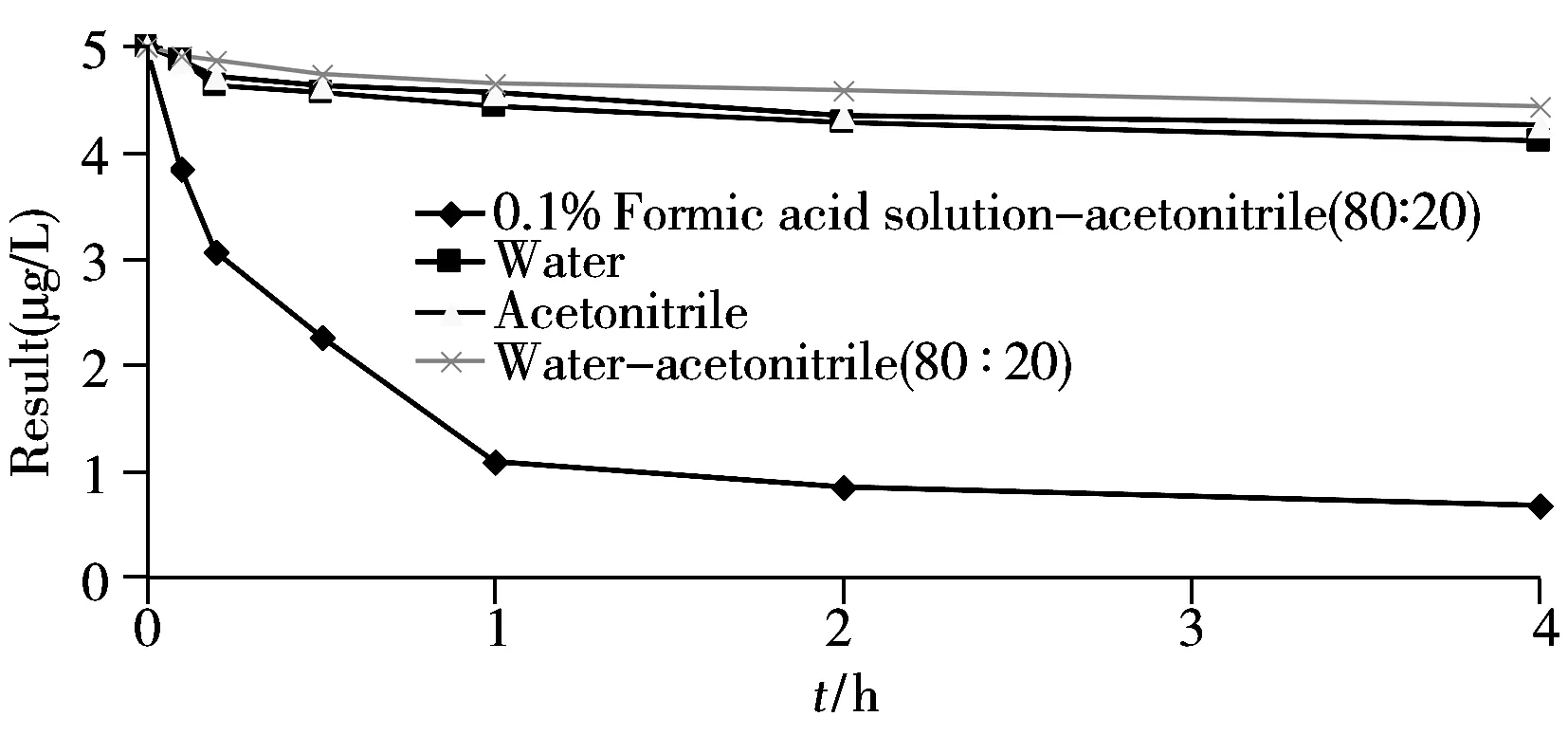
图2 N-甲基邻苯二胺(5.0 μg/L)在不同配制溶液中的降解曲线Fig.2 Degradation curve of N-methyl-o-phenylenediaie(5.0 μg/L)in different solution
2 结果与讨论
2.1 配制溶液的选择
N-甲基邻苯二胺含有1个氨基,溶解后呈弱碱性,在酸性环境下易发生降解。因此,本研究考察了不同配制溶液对N-甲基邻苯二胺降解速度的影响,分别采用初始流动相(0.1%甲酸水溶液-乙腈,80∶20)、水、乙腈和水-乙腈(80∶20)配制浓度为5.0 μg/L的N-甲基邻苯二胺对照品溶液,放置0,0.1,0.2,0.5,1,2,4 h后上机检测,发现N-甲基邻苯二胺在0.1%甲酸水溶液-乙腈中的降解速度较快,降解幅度较大,而在水、乙腈和水-乙腈(80∶20)中的降解速度较慢,降解幅度较小(见图2)。进一步研究发现,替米沙坦在水中的溶解度非常差,不利于供试品溶液的配制;乙腈对替米沙坦的溶解性虽较好,但配制后N-甲基邻苯二胺和替米沙坦主峰的峰形均不好,而采用水-乙腈溶液(80∶20)配制后的峰形较好,替米沙坦的溶解度也较好,保证了样品与对照品配制溶液的一致性,故本文选用水-乙腈溶液(80∶20)作为N-甲基邻苯二胺标准溶液和替米沙坦供试液的配制溶液。
2.2 色谱条件的选择
N-甲基邻苯二胺的极性非常大,出峰时间较早,而替米沙坦的极性偏小,出峰相对较晚,二者的分离难度不大,且N-甲基邻苯二胺在主峰之前出峰,基本不受替米沙坦的干扰影响。本文在相近的色谱条件下,考察了Agilent Eclipse XDB-C18色谱柱(3.5 μm,2.1 mm×100 mm)、Thermo Hypersil-C18色谱柱(1.9 μm,2.1 mm×100 mm)和Agilent Extend-C18(1.8 μm,2.1 mm×50 mm) 3种色谱柱的分离情况。结果显示,上述3种色谱柱均能实现替米沙坦主峰和N-甲基邻苯二胺的有效分离,且N-甲基邻苯二胺的质谱峰形也较好。但考虑到Thermo Hypersil-C18和Agilent Extend-C18色谱柱的柱容量均较小,检测的进样量为10 μL,多次进样后易残留替米沙坦,影响检测结果。而Agilent Eclipse XDB-C18色谱柱的内径为3.5 μm,柱容量较大,主峰基本不会残留,故本文选用Agilent Eclipse XDB-C18色谱柱进行分离。
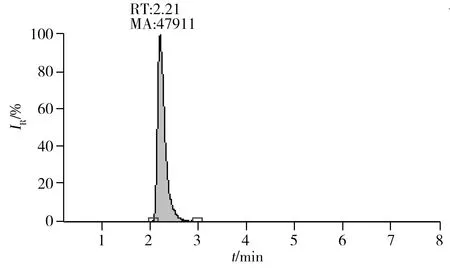
图3 N-甲基邻苯二胺标准的SRM色谱图Fig.3 SRM chromatogram of N-methyl-o-phenylenediamine
2.3 质谱条件的选择
采用电喷雾离子源,在正离子模式下,对1.0 mg/L的N-甲基邻苯二胺标准溶液进行母离子全扫描,确定其分子离子[M+H]+峰(m/z123.1);对分子离子峰进行碎裂并扫描子离子,得到二级质谱图,筛选响应值较高、基线噪音低的离子(m/z107.6)作为监测离子。采用选择反应检测(SRM)模式采集数据,驻留时间设定在100 ms,优化各离子的碰撞能,得到最佳质谱条件如“1.4”所述。在优化条件下,得N-甲基邻苯二胺标准的SRM色谱图(图3)。
2.4 方法的线性范围、检出限及定量下限
以水-乙腈(80∶20)配制0.0,2.0,5.0,10,20,50 μg/L的N-甲基邻苯二胺标准系列溶液分别进行测定,以定量离子峰面积对质量浓度进行线性回归计算。结果表明,N-甲基邻苯二胺在0.0~50 μg/L范围内线性关系良好(r>0.99);以质谱3倍基线噪音计算检出限(S/N=3)为0.50 μg/L,以质谱10倍基线噪音计算定量下限(S/N=10)为2.0 μg/L,完全满足依照欧盟基因毒性杂质指导原则计算的5.0 μg/L浓度的杂质限度。
2.5 回收率实验
采用不含N-甲基邻苯二胺的替米沙坦精品为空白基质,进行不同浓度的加标回收实验时发现,各加标浓度下的回收率均较低;且在精密度试验时发现,随着检测时间的延长回收率逐渐降低,可能是N-甲基邻苯二胺在替米沙坦溶液中发生了降解。研究发现,替米沙坦具有羧酸结构,其样品溶液呈弱酸性,而N-甲基邻苯二胺在酸性条件下会发生降解。进一步做降解试验发现,N-甲基邻苯二胺在替米沙坦溶液中呈明显的降解趋势,但降解的速率较慢(见表1),不同浓度的N-甲基邻苯二胺在1 h内的降解率均不超过5%,若能在添加实验时现配现测,基本不会影响回收率效果。本文也尝试将替米沙坦溶液调节pH值至中性,回收率可得到明显改善,但操作较为繁琐,且相应的对照品溶液也需调节pH值,不利于实际操作。因此,本文采用不含N-甲基邻苯二胺的替米沙坦精品为空白基质,添加了80%限度、100%限度、120%限度浓度的N-甲基邻苯二胺分别做回收率和精密度试验,每个浓度设3个平行,且所有添加样品均在溶液配制1 h内进行检测,得到平均回收率为85.7%~94.3%,相对标准偏差(RSD)为1.9%~2.9%(见表2),表明方法的回收率良好,完全可以满足检测要求。
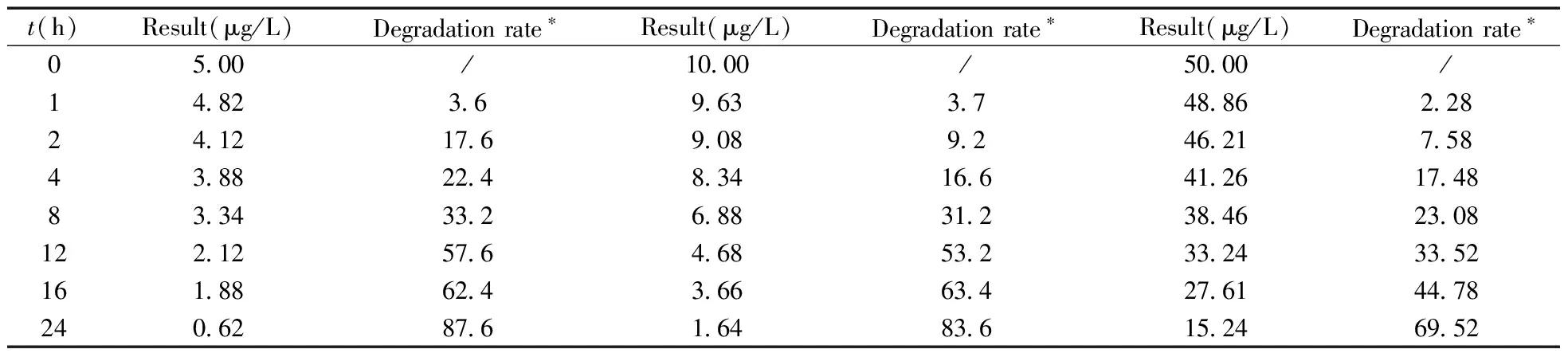
表1 不同添加浓度的N-甲基邻苯二胺在替米沙坦溶液中的降解率Table 1 Degradation rate of N-methyl-o-phenylenediaine in telmisartan at different spiked levels /%
*degradation rate =(CA-CF)/CA×100%,CA:added of N-methyl-o-phenylenediaine,CF:founded of N-methyl-o-phenyl-enediaine at different time points
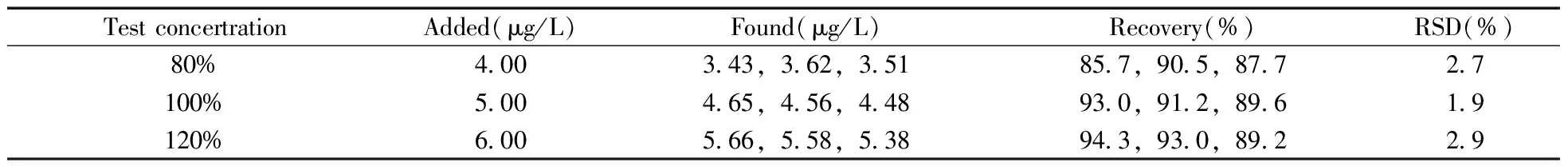
表2 替米沙坦中添加N-甲基邻苯二胺的回收率及相对标准偏差Table 2 Recoveries and RSDs of N-methyl-o-phenylenediamine in telmisartan
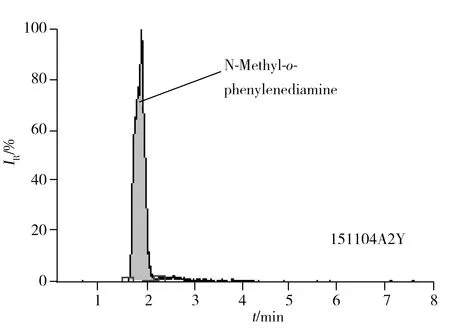
图4 替米沙坦成品中含N-甲基邻苯二胺的阳性样品色谱图(151104A2Y)Fig.4 Chromatogram of positive samples with N-methyl-o-pheny-lenediamine in telmisartan(151104A2Y)
2.6 实际应用
将所建方法应用于检测合成的药物小试3批、中试3批及工艺验证3批共27份样品,所有样品均在配制1 h内检测,共检出6批含有N-甲基邻苯二胺残留,检出结果分别为2.60 mg/kg(151216A2J),3.21 mg/kg(151216B1J),49.42 mg/kg(151216B1Y),12.30 mg/kg(151104A2Y),6.46 mg/kg(151104B2Y),7.73 mg/kg(151104B3Y),其中有4批样品超过限度要求,其他样品均未检出,151104A2Y阳性样品谱图见图4。
3 结 论
本文首次建立了高效液相色谱-四极杆/线性离子阱质谱测定替米沙坦药物中基因毒性杂质N-甲基邻苯二胺残留量的分析方法。该方法灵敏度高、分离度好,回收率、精密度及定量下限均能满足基因毒性杂质的限度要求,是检测替米沙坦药物中N-甲基邻苯二胺残留量的有效方法。将所建方法用于检测合成的替米沙坦药物样品,共检出6批含有N-甲基邻苯二胺残留,方法有效地指导了替米沙坦合成反应的进行,并为项目的研发过程起到了良好的监督作用。
[1] Yang X T,Gao H L,Liu Y J,Zou F,Qu F J.Chin.J.Pharm.(杨秀婷,高华露,柳玉杰,邹枫,曲福军.中国药师),2015,18(9):1465-1468.
[2] Lü Y P,Zhang J,Zhang A H,Zhang J,Wang Y H,Duan K D.Chin.J.Med.Guide(吕亚萍,张军,张爱华,张杰,王英海,段克东.中国医药导刊),2015,17(7):727-728.
[3] Lan X Y,Xu S H.Chin.J.Med.Herald(兰行远,徐尚华.中国医药导报),2015,12(33):147-150.
[4] European Medicines Agency.Guideline on the Limits of Genotoxic Impurities[EB/OL].http://www.ema.eu- ropa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002903.pdf,2006-06-28/2013-12-13.
[5] Munro I C,Renwick A G,Danielewska-Nikiei B.Toxicol.Lett.,2008,180(2):151-156.
[6] Yin D R.Chin.J.HealthLab.Technol.(殷德荣.中国卫生检验杂志),2013,23(11):2549-2552.
[7] Zhao X M,Wang Z L,Wang H P,Lu Z Y,Shao Y,Yao J W.J.Environ.Sci.Technol.(赵学梅,王志良,汪海萍,陆朝阳,邵燕,姚继伟.环境科技),2010,23(6):46-47.
[8] Shen X,Zhou Y Y.J.AnhuiNormalUniv.:Nat.Sci.(沈新,周运友 .安徽师范大学学报:自然科学版),2009,32(6):561-564.
[9] Ji Z P,Wang J,Hu X Y.Chin.J.Anal.Chem.(嵇正平,王俊,胡效亚.分析化学),2009,10(37):B139.
[10] Wang J C,Ye C,Wang D H.ThesisCollectionofZhejiangProvinceAnalysisandTestingAcademicReport.Hangzhou:Zhejiang Province Analysis and Testing Academic Report(王金朝,叶春,王丹华.浙江省分析测试学术报告会论文集 .杭州:浙江省分析测试学术报告会),2007.
[11] Venugopal N,Vijaya Bhaskar Reddy A,Gangadhar Reddy K,Madhavi V,Madhavi G.J.Pharm.Biomed.Anal.,2012,70:592-597.
[12] Vijaya Bhaskar Reddy A,Venugopal N,Madhavi G,Gangadhar Reddy K,Madhavi V.J.Pharm.Biomed.Anal.,2013,84:84-89.
[13] Liu L Y,Zhang Q R,Sun L F,Wang M M,Guo W M.Chin.J.Pharm.(刘立云,张倩如,孙连福,王曼曼,郭文敏.中国药师),2014,17(8):1274-1276.
[14] Ding Y M,Wu J,Lu C X,Cui P,Wang D C.Chin.J.Pharm.(丁逸梅,吴娟,陆春晓,崔萍,王德才.中国医药工业杂志),2014,45(11):1069-1071.
[15] Liu J H,Zhong Y N,Xiong Y,He W T.Chin.J.Pharm.Anal.(刘俊华,钟雅妮,熊渊,赫涡涛.药物分析杂志),2013,33(7):1168-1170.
[16] Zhang L,Huang L N,Li Q R,Yang W H.Chin.J.Pharm.Anal.(张莉,黄丽娜,黎其荣,杨伟涵.药物分析杂志),2015,35(2):317-319.
[17] Zhang H W,Jian H M,Lin L M,Chen L Z,Liang C Z,Yuan T,Tang Z X,Cai X,Qin L Y.J.Instrum.Anal.(张鸿伟,简慧敏,林黎明,陈亮珍,梁成珠,袁涛,汤志旭,蔡雪,秦良勇.分析测试学报),2012,31(7):763-770.
[18] Yang J J,Wu S Q,Tong L,Song S L,Tian Q.J.Instrum.Anal.(杨佳佳,吴淑琪,佟玲,宋淑玲,田芹.分析测试学报),2011,30(4):374-380.
[19] Liu J,Zhang M,Wan Y,Wu H M,Hou Y N,Zheng L M,Yi Y X.Chin.J.Anal.Chem.(刘佳,张朦,万有,吴红梅,侯艳宁,郑乐民,尹玉新.分析化学),2014,43(8):1118-1124.
[20] Ruan X L,Zhang A H,Wu C,Rong W F,Huang S L.J.Instrum.Anal.(阮小林,张爱华,吴川,戎伟丰,黄淑莲.分析测试学报),2011,30(1):72-75.
[21] Luo Y Y,Liu J X,Liu X H,Lan C W,Hou Y,Ma Y,Xu L.J.Instrum.Anal.(罗益远,刘娟秀,刘训红,兰才武,侯娅,马阳,徐力.分析测试学报),2015,34(5):519-524.
[22] Zhou Y,Zhao Y G,Zhang B B,Zhang Y,Chen G S.Chin.J.Anal.Chem.(周岩,赵永刚,张蓓蓓,章勇,陈国松.分析化学),2014,42(3):367-374.
Determination of N-Methyl-o-phenylenediamine in Telmisartan by HPLC-QTRAP-MS/MS
ZHOU Chang-peng*,WANG Dong-wu,XU Jie,ZHENG Wen-feng,LIANG Xiao-yan
(National Enterprise Technology Center of Disha Pharmaceutical Group,Weihai 264205,China)
A high performance liquid chromatography-quadrupole ion mass spectrometric(HPLC-QTRAP-MS/MS) method was developed for the determination of N-Methyl-o-phenylenediamine residues in telmisartan.After extracted with water-acetonitrile(80∶20,by volume),the telmisartan samples were separated on an Agilent Eclipse XDB-C18column(3.5 μm,2.1 mm×100 mm) with a mobile phase of 0.1% formic acid solution-acetonitrile.The electrospray was operated in the positive mode and the samples were monitored under the select reaction monitoring(SRM) mode.As a result,the calibration curves of N-Methyl-o-phenylenediamine showed a good linearity(r>0.99) in the range of 2.0-50 μg/L.The limits of detection(S/N=3) was 0.5 μg/L ,and the limits of quantitation(S/N=10) was 2.0 μg/L.The method has the advantages of simple operation,high sensitivity,good reproducibility,and could be used for the detection of N-Methyl-o-phenylenediamine residues in telmisartan.
high performance liquid chromatography-quadrupole ion mass spectrometry(HPLC-QTRAP-MS/MS);N-methyl-o-phenylenediamine;telmisartan
2016-09-09;
2016-09-28
10.3969/j.issn.1004-4957.2017.02.019
O657.63;TQ460.726
A
1004-4957(2017)02-0262-05
*通讯作者:周长朋,工程师,研究方向:理化检测及药物分析,Tel:0631-3857373,E-mail:20019568@qq.com
