膝骨关节炎脂肪垫对关节软骨细胞的影响
2017-03-13瞿燕萍高明霞夏鹏张婷婷程凯林强李雪萍
瞿燕萍,高明霞,夏鹏,张婷婷,程凯,林强,李雪萍
脂肪组织不仅是能量储存器官,还是重要的内分泌器官,它可分泌多种脂肪因子和细胞因子调控全身代谢以维持机体健康[1]。由过多的脂肪组织堆积而成的肥胖可引起多种疾病,如2型糖尿病、高血压病、血脂异常和骨代谢性疾病,后者包括骨质疏松症和骨关节炎(Osteoarthritis, OA)[2]。OA是一种以关节软骨退变,骨赘形成,滑膜炎症及软骨下骨硬化为特征的退行性关节疾病[3]。OA是最常见的风湿性疾病和身体残疾的主要原因,可严重降低中老年人群的生活质量,并且随着社会人口老龄化、肥胖以及久坐等危险因素的增加,OA的发病率逐年升高[4]。研究表明由脂肪组织分泌的脂肪因子作用于关节软骨组织可促进关节炎症、抑制软骨基质合成及刺激软骨下骨重塑[5-7],还可通过上调和活化蛋白水解酶对软骨代谢起促分解作用[8-9]。脂肪因子包括瘦素、脂联素、抵抗素以及内脂素等,其中,瘦素是最早被发现且为目前研究最多的脂肪因子,它可抑制食物摄入以及刺激能量消耗,并参与调节免疫反应和炎性疾病[10-11]。研究表明,脂肪因子在OA的发生及发展中起重要作用[12-13]。膝髌下脂肪垫(Infrapatellar Fat Pad, IPFP)是膝关节囊内滑膜外与软骨相邻的脂肪组织,是膝关节局部脂质和脂肪因子的来源,研究证明与IPFP有关的代谢作用可引起或加重软骨损害以及关节炎症和疼痛,此外,IPFP分泌的脂肪因子还能刺激滑膜中促炎性细胞因子和蛋白水解酶的表达[6, 14]。本研究通过制作OA膝脂肪垫培养液(Fat Conditioned Medium, FCM)并与正常及OA软骨细胞共培养,观察FCM对正常及OA软骨细胞的影响,旨在探讨OA膝脂肪垫对关节软骨的作用。
1 材料与方法
1.1 材料 2015年10月~2016年7月收集的24例膝软骨和膝脂肪垫均由骨科手术室提供,12例为女性膝关节急性外伤手术患者,12例为女性膝OA患者行关节置换术后,均排除其他膝关节病史,年龄均在40~55岁之间。所有实验方案均得到南京医科大学附属南京医院伦理委员会批准。主要器材及试剂包括胎牛血清、Ⅱ型胶原酶(Giboco公司),胰蛋白酶、高糖不完全达尔伯克改良伊格尔培养基(Dulbecco's Modified Eagle's Medium, DMEM)、全蛋白提取试剂盒、免疫组化试剂盒、油红染料(南京凯基生物有限公司),瘦素抗体(武汉博士德生物工程研究所),Ⅱ型胶原(Type Ⅱ Collagen, COL2)抗体、聚蛋白多糖(Aggrecan, Acan)抗体、基质金属蛋白酶(Matrixmetalloproteinase, MMP)-13抗体(Abcam 公司),Western电泳仪(Bio-Rad,美国,型号164-5051),电泳仪及湿式电转移槽(美国Bio-Rad公司)。
1.2 方法 ①膝脂肪垫HE染色:将正常及OA膝脂肪垫浸入福尔马林溶液,经苏木精-伊红染色,进行病理分析。②瘦素免疫组化染色:将正常及OA膝脂肪垫分别制作石蜡切片,做免疫组织化学染色,即用型山羊血清,瘦素抗体(1∶100稀释),加酶标二抗,DAB溶液显色,玻片在光学显微镜下观察组织中瘦素蛋白的表达情况。③制作FCM[15]:将OA膝脂肪垫用PBS液冲洗2~3遍,去除纤维结缔组织及血管,剪切至小碎块大约50mg,再用PBS液冲洗2遍及无血清的高糖DMEM培养基冲洗1遍。加入完全高糖DMEM培养基(含5%胎牛血清+链霉素100μg/ml+青霉素100IU/ml)置于37℃恒温箱中24h,随后替换为无血清的高糖DMEM培养基(1ml/0.3g 脂肪组织)于37℃恒温箱中72h。最后用200目分子筛过滤脂肪垫并收集培养液,置于-80℃备用。④软骨细胞分离与培养:于急性外伤手术标本的表面完整处提取正常软骨细胞,于OA膝关节置换术标本提取OA软骨细胞。将膝软骨用PBS液冲洗并剪切至1.0mm3的小碎片,移入15ml离心管并加入2ml 0.25%胰蛋白酶消化30min,再加入3ml 0.25%Ⅱ型胶原(TypeⅡ Collagen, COL2)酶,于37℃恒温箱中消化4h,每半小时振荡一次,加入3ml培养基终止消化,予1200rpm/min离心10min,弃上清,加入完全高糖DMEM培养基(含10%的胎牛血清)吹打均匀后种植于培养瓶内,置于5%CO2、95%空气37℃恒温培养箱中。每天用光学显微镜观察细胞生长情况,待细胞铺满瓶底80%~90%时传代。⑤实验分组与干预:将第2代正常软骨细胞采用随机数字法分为正常组和正常+FCM培养组,第2代OA软骨细胞采用随机数字法分为OA组和OA+FCM培养组,其中在正常组和OA组中加入完全高糖DMEM培养基,在正常+FCM培养组和OA+FCM培养组中加入FCM,每天于显微镜下观察,第14天将各组细胞予COL2免疫组化染色及油红O染色,并收集各组细胞予Western blot检测。⑥COL2免疫组化染色:将爬有细胞的盖玻片用4%的多聚甲醛固定15min,COL2抗体(1∶100稀释),具体实验步骤参考瘦素免疫组织化学染色。光学显微镜下可见软骨细胞胞核深染,胞质为大量COL2染色,呈棕色。⑦油红O染色:将软骨细胞用PBS冲洗2次,10%的福尔马林固定15min,60%的异丙醇漂洗30s,PBS冲洗2次,油红O染色2h,倾去染液,流水冲洗,苏木精染色1min,倾去苏木精,流水冲洗,中性树胶+盖玻片固定,脂滴被油红染成橘红色,细胞核被苏木精染成蓝色。⑧免疫印迹法检测(Western blot):采用聚丙烯酰胺凝胶电泳,湿式转移电泳槽转膜,聚偏二氟乙烯膜在5%的牛血清白蛋白中常温震荡封闭2h,分别加Ⅱ型胶原(TypeⅡ Collagen, COL2)抗体、聚蛋白多糖(Aggrecan, Acan)抗体、基质金属蛋白酶(Matrix Metalloproteinases, MMP)-13抗体和磷酸甘油醛脱氢酶(Glyceraldehyde-3-Phosphate Dehydrogenase, GAPDH)抗体(1∶1000稀释),4℃孵育过夜,加二抗,常温孵育2h,加显影液,采用机器曝光。Image pro plus 6.0 图像分析软件对各组蛋白电泳条带行COL2、Acan、MMP-13、GAPDH的灰度分析。

2 结果
2.1 脂肪垫组织病理学 正常膝脂肪垫内可见大量呈空泡状的脂肪细胞(放大400倍),彼此相连,见图1a;OA膝脂肪垫内亦可见大量脂肪细胞,脂肪细胞间质有较多巨噬细胞、淋巴细胞和浆细胞等炎性细胞浸润,呈蓝色颗粒,见图1b。

图1 膝脂肪垫HE染色(×400)
注:a.正常膝脂肪垫;b.OA膝脂肪垫
2.2 瘦素免疫组织化学染色 正常膝脂肪垫瘦素免疫组织化学染色呈阳性反应(0.63±0.02),见图2a;OA膝脂肪垫瘦素免疫反应较正常组增多(0.71±0.02),见图2b。

图2 正常及OA膝脂肪垫瘦素免疫组织化学染色(×200)
注:a.正常膝脂肪垫;b.OA膝脂肪垫;图中黑色箭头示脂肪细胞核,红色箭头示瘦素阳性
2.3 COL2免疫组织化学染色 OA组软骨细胞内的COL2免疫组化染色平均吸光度比正常组显著降低(P<0.05)。与OA组相比,正常+FCM组软骨细胞内的COL2免疫组化染色平均吸光度无明显差异(P>0.05),而OA+FCM组显著降低(P<0.05)。与正常+FCM组相比,OA+FCM组软骨细胞内的COL2免疫组化染色平均吸光度显著降低(P<0.05)。见表1,图3。
2.4 油红O染色 OA组软骨细胞内的油红O染色平均吸光度与正常组相比差异无统计学意义。与OA组相比,正常+FCM组和OA+FCM组软骨细胞内的油红O染色平均吸光度均有增高,但OA+FCM组增高更为显著(P<0.05)。见表1,图4。

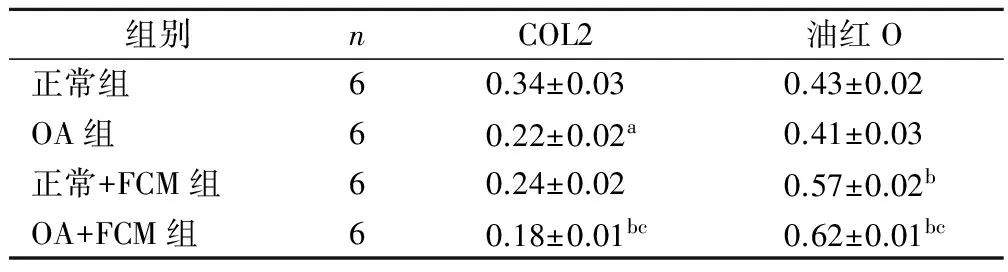
组别nCOL2油红O正常组60.34±0.030.43±0.02OA组60.22±0.02a0.41±0.03正常+FCM组60.24±0.020.57±0.02bOA+FCM组60.18±0.01bc0.62±0.01bc
与正常组比较,aP<0.05;与OA组比较,bP<0.05;与正常+FCM组相比,cP<0.05
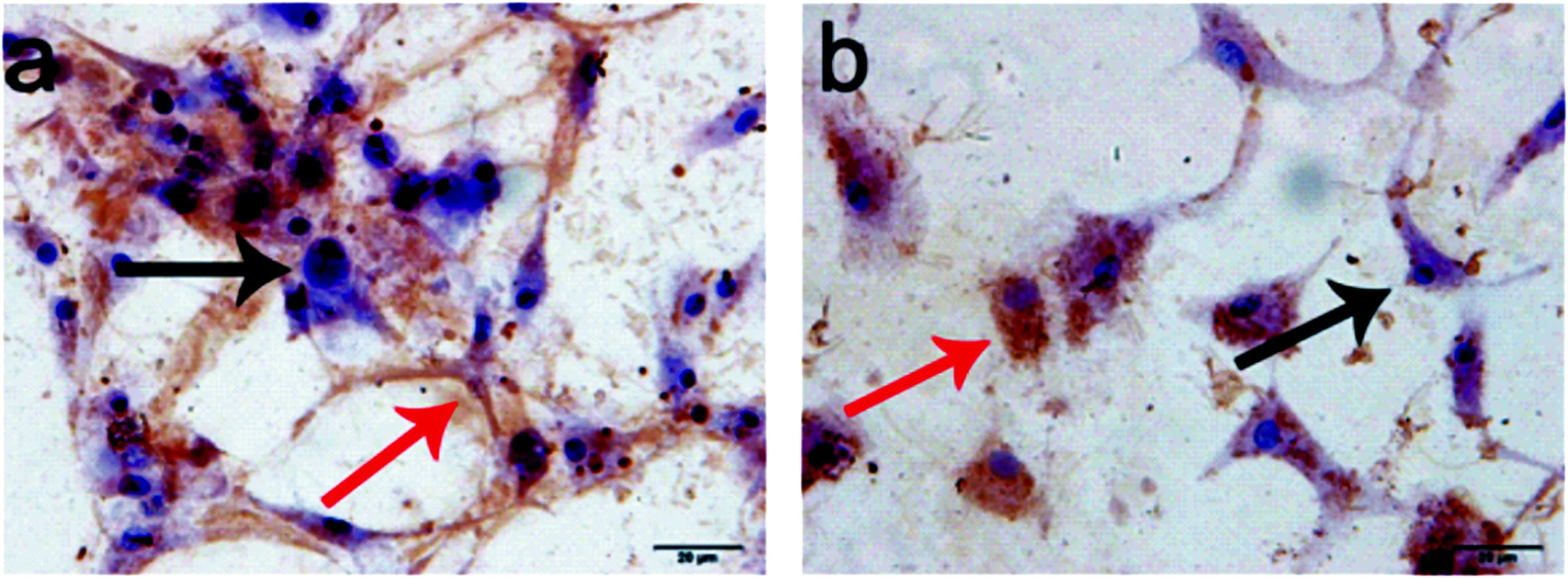

图3 4组软骨细胞COL2免疫组化染色(×200)
注:a.正常组;b.OA组;c.正常+FCM组;d.OA+FCM组;图中黑色箭头示软骨细胞核,红色箭头示COL2免疫组化染色阳性


图4 4组软骨细胞油红O染色(×200)
注:a.正常组;b.OA组;c.正常+FCM组;d.OA+FCM组;图中黑色箭头示软骨细胞核,红色箭头示细胞内油红O染色阳性
2.5 各组软骨细胞中COL2、Acan和MMP-13蛋白Western检测表达水平比较 OA组COL2、Acan的表达水平低于正常组(P<0.05),而MMP-13的表达高于正常组(P<0.05)。与OA组相比,正常+FCM组3种蛋白的表达水平均无明显差别;相较于正常组及正常+FCM组,OA+FCM组COL2、Acan表达水平降低(均P<0.05),而MMP-13的表达水平增高(均P<0.05),见表2,图5。

组别nCOL2AcanMMP⁃13正常组60.49±0.030.63±0.040.05±0.01OA组60.28±0.06a0.26±0.05a0.34±0.05a正常+FCM组60.24±0.090.24±0.060.31±0.08OA+FCM组60.07±0.04bc0.10±0.03bc0.45±0.03bc
与正常组比较,aP<0.05;与OA组比较,bP<0.05;与正常+FCM组相比,cP<0.05

图5 4组软骨细胞各目标蛋白电泳图
3 讨论
关节软骨是由少量软骨细胞和大量细胞外基质组成。软骨细胞是软骨组织内唯一的细胞类型,其细胞形态、增殖能力及分泌COL2的量可反映软骨细胞的活性[16]。细胞外基质主要由软骨细胞分泌,包括胶原(主要为COL2)和Acan,其中COL2占细胞外基质总量的80%~90%,是软骨细胞的特征性指标。COL2可形成纤维网络结构使软骨具有抗张强度,Acan主要保持软骨的弹性和黏度,其中COL2的降解是不可逆的[17],由此可见COL2和Acan在维持软骨的完整与功能中起重要作用。
近年来,越来越多的研究将脂肪组织与软骨退行性变联系在一起[18-19]。实验证明,采用高脂饮食诱导的肥胖小鼠,其膝软骨损伤水平与机械应力引起的软骨损伤相似[20],并且通过饮食诱导的肥胖还可显著增加小鼠创伤后关节炎的严重程度[21]。由此可见,脂肪组织可通过代谢途径对软骨产生破坏作用。脂肪组织主要由脂肪细胞构成,其细胞间质有炎性细胞浸润如巨噬细胞、淋巴细胞和浆细胞等[22]。脂肪细胞可分泌众多脂肪因子包括瘦素、脂联素、内脂素和抵抗素等,脂肪因子能以自分泌或旁分泌双重方式刺激软骨细胞。
在软骨细胞中,瘦素通过结合软骨细胞表面瘦素受体活化细胞内促分裂原活化蛋白激酶(Mitogen Activated Protein Kinase, MAPK)信号通路,进而促进软骨细胞MMP-13的表达以及诱导软骨胶原释放,对软骨基质代谢起促分解作用[23],MMPs是降解细胞外基质最重要的蛋白水解系统,其中MMP-13是COL2最有效地蛋白水解酶[24]。OA软骨的显著受损区域相比未受损区域可表达更多的瘦素受体,并且瘦素在OA滑膜液中的表达水平显著高于血清[25]。膝脂肪垫是位于膝关节囊内、滑膜外并与软骨相邻的脂肪组织,是膝关节脂肪因子的局部来源,研究指出脂肪垫分泌的脂肪因子脂联素和内脂素显著高于皮下脂肪组织[26],并且随着年龄或体重指数的增加,脂肪垫内的脂肪细胞和浸润的淋巴细胞数量会增多,增多的脂肪细胞将进一步提高膝关节内脂肪因子的水平[27-29]。此外,实验证明OA患者的脂肪垫相比正常脂肪垫可分泌更多的脂肪因子[30-31],OA患者的脂肪垫相比其皮下脂肪可产生更多的炎症细胞[32],并且晚期OA患者的脂肪垫相比早期OA患者的脂肪垫可表达更多的炎性因子[33]。由此可见,脂肪垫作为膝关节组织的一部分可以调节膝关节炎症和软骨降解反应。
本研究通过将正常膝脂肪垫与OA膝脂肪垫行HE染色提示OA脂肪垫可产生更多的炎症细胞,并将正常膝脂肪垫与OA膝脂肪垫行瘦素免疫组织化学染色提示OA脂肪垫呈现更多的瘦素免疫反应物,由此可见,OA脂肪垫可表达更多的炎性因子,第2代正常软骨细胞生长快,而第2代OA软骨细胞生长速度较慢,并且油红O染色结果显示正常及OA软骨细胞内均呈现少量脂质成分,脂滴呈橘红色,2组间的平均吸光度无明显差异(P>0.05)。加入FCM后发现,正常+FCM培养组软骨细胞以多边形为主,其中夹杂不少树突样细胞,生长速度慢,并且细胞内出现少量大小不一的透明空泡,随着时间的增加,空泡数量也逐渐增多,此时软骨细胞仍为多边形或星形,经油红O染色后细胞内出现较多红染颗粒,证明镜下所见细胞内的透明空泡为脂滴,正常+FCM培养组的油红O染色平均吸光度比OA组显著增高(P<0.05),提示FCM培养下的正常软骨细胞内脂滴含量增多,软骨细胞发生脂肪样变。OA+FCM培养组软骨细胞主要呈树突样,生长速度缓慢,镜下细胞内呈现大量透明空泡,经油红O染色后细胞内出现大量红染颗粒,其平均吸光度比正常+FCM组显著增高(P<0.05),说明FCM培养下的OA软骨细胞脂滴明显增加,OA+FCM培养组软骨细胞发生脂肪样变较正常+FCM培养组软骨细胞更严重,进一步证明OA膝脂肪垫能对软骨细胞产生破坏作用,而对OA软骨细胞的破坏更严重。
由COL2免疫组化结果可见,OA脂肪垫通过分泌脂肪因子对软骨细胞基质代谢起促分解作用,结合上述油红O染色结果进一步证实了OA脂肪垫能对软骨产生破坏作用。本研究进一步通过Western Blot检测证明脂肪因子在软骨退行性变过程中可通过代谢途径提高软骨细胞MMP-13的表达进而促进软骨细胞外基质的降解,最终引起软骨损伤。
该实验的创新性表现在,通过将OA膝脂肪垫培养液与正常及OA软骨细胞共培养证明脂肪因子对软骨细胞产生破坏作用,并且对OA软骨细胞产生更大的破坏作用。此外,本实验首次证明与OA膝脂肪垫培养液共培养的正常及OA软骨细胞可发生脂肪样变,并且OA软骨细胞发生脂肪样变更为显著。然而,为何软骨细胞会在与脂肪垫培养液共培养的状态下会发生脂肪样变,需要未来进一步的实验探讨。
[1] Booth A, Magnuson A, Fouts J, et al. Adipose tissue: an endocrine organ playing a role in metabolic regulation[J]. Horm Mol Biol Clin Investig, 2016, 26(1): 25-42.
[2] Lee R, Kean WF. Obesity and knee osteoarthritis[J]. Inflammopharmacology, 2012, 20(2): 53-58.
[3] Xia P, Shen S, Lin Q, et al. Low-Intensity Pulsed Ultrasound Treatment at an Early Osteoarthritis Stage Protects Rabbit Cartilage From Damage via the Integrin/Focal Adhesion Kinase/Mitogen-Activated Protein Kinase Signaling Pathway[J]. J Ultrasound Med, 2015, 34(11): 1991-1999.
[4] Sartori-Cintra AR, Aikawa P, Cintra DE. Obesity versus osteoarthritis: beyond the mechanical overload[J]. Einstein (Sao Paulo), 2014, 12(3): 374-379.
[5] Iannone F, Lapadula G. Obesity and inflammation——targets for OA therapy[J]. Curr Drug Targets, 2010, 11(5): 586-598.
[6] Santangelo KS, Radakovich LB, Fouts J, et al. Pathophysiology of obesity on knee joint homeostasis: contributions of the infrapatellar fat pad[J]. Horm Mol Biol Clin Investig, 2016, 26(2): 97-108.
[7] Sellam J, Berenbaum F. Is osteoarthritis a metabolic disease[J]? Joint Bone Spine, 2013, 80(6): 568-573.
[8] Gomez R, Lago F, Gomez-Reino J, et al. Adipokines in the skeleton: influence on cartilage function and joint degenerative diseases[J]. J Mol Endocrinol, 2009, 43(1): 11-18.
[9] Yaykasli KO, Hatipoglu OF, Yaykasli E, et al. Leptin induces ADAMTS-4, ADAMTS-5, and ADAMTS-9 genes expression by mitogen-activated protein kinases and NF-kB signaling pathways in human chondrocytes[J]. Cell Biol Int, 2015, 39(1): 104-112.
[10] Matarese G. Leptin and the immune system: how nutritional status influences the immune response[J]. Eur Cytokine Netw, 2000, 11(1): 7-14.
[11] Lam QL, Lu L. Role of leptin in immunity[J]. Cell Mol Immunol, 2007, 4(1): 1-13.
[12] Koskinen A, Juslin S, Nieminen R, et al. Adiponectin associates with markers of cartilage degradation in osteoarthritis and induces production of proinflammatory and catabolic factors through mitogen-activated protein kinase pathways[J]. Arthritis Res Ther, 2011, 13(6): R184-R184.
[13] Gomez R, Lago F, Gomez-Reino J, et al. Adipokines in the skeleton: influence on cartilage function and joint degenerative diseases[J]. J Mol Endocrinol, 2009, 43(1): 11-18.
[14] Clockaerts S, Bastiaansen-Jenniskens YM, Runhaar J, et al. The infrapatellar fat pad should be considered as an active osteoarthritic joint tissue: a narrative review[J]. Osteoarthritis Cartilage, 2010, 18(7): 876-882.
[15] Hui W, Litherland GJ, Elias MS, et al. Leptin produced by joint white adipose tissue induces cartilage degradation via upregulation and activation of matrix metalloproteinases[J]. Ann Rheum Dis, 2012, 71(3): 455-462.
[16] Schindler OS. Current concepts of articular cartilage repair[J]. Acta Orthop Belg, 2011, 77(6): 709-726.
[17] Karsdal MA, Madsen SH, Christiansen C, et al. Cartilage degradation is fully reversible in the presence of aggrecanase but not matrix metalloproteinase activity[J]. Arthritis Res Ther, 2008, 10(3): R63.
[18] Thijssen E, Caam A, Kraan PM. Obesity and osteoarthritis, more than just wear and tear: pivotal roles for inflamed adipose tissue and dyslipidaemia in obesity-induced osteoarthritis[J]. Rheumatology (Oxford), 2015, 54(4): 588-600.
[19] Zhuo Q, Yang W, Chen J, et al. Metabolic syndrome meets osteoarthritis[J]. Nat Rev Rheumatol, 2012, 8(12): 729-737.
[20] Collins KH, Reimer RA, Seerattan RA, et al. Using diet-induced obesity to understand a metabolic subtype of osteoarthritis in rats[J]. Osteoarthritis Cartilage, 2015, 23(6): 957-965.
[21] Louer CR, Furman BD, Huebner JL, et al. Diet-induced obesity significantly increases the severity of posttraumatic arthritis in mice[J]. Arthritis Rheum, 2012, 64(10): 3220-3230.
[22] Iwata M, Ochi H, Hara Y, et al. Initial responses of articular tissues in a murine high-fat diet-induced osteoarthritis model: pivotal role of the IPFP as a cytokine fountain[J]. PLoS One, 2013, 8(4): e60706.doi: 10.1371/journal.pone.0060706. Print 2013.
[23] Richter M, Trzeciak T, Owecki M, et al. The role of adipocytokines in the pathogenesis of knee joint osteoarthritis[J]. Int Orthop, 2015, 39(6): 1211-1217.
[24] Burrage PS, Mix KS, Brinckerhoff CE. Matrix metalloproteinases: role in arthritis[J]. Front Biosci, 2006, 11: 529-543.
[25] Simopoulou T, Malizos KN, Iliopoulos D, et al. Differential expression of leptin and leptin's receptor isoform (Ob-Rb) mRNA between advanced and minimally affected osteoarthritic cartilage; effect on cartilage metabolism[J]. Osteoarthritis Cartilage, 2007, 15(8): 872-883.
[26] Klein-Wieringa IR, Kloppenburg M, Bastiaansen-Jenniskens YM, et al. The infrapatellar fat pad of patients with osteoarthritis has an inflammatory phenotype[J]. Ann Rheum Dis, 2011, 70(5): 851-857.
[27] Duran S, Aksahin E, Kocadal O, et al. Effects of body mass index, infrapatellar fat pad volume and age on patellar cartilage defect[J]. Acta Orthop Belg, 2015, 81(1): 41-46.
[28] Chuckpaiwong B, Charles HC, Kraus VB, et al. Age-associated increases in the size of the infrapatellar fat pad in knee osteoarthritis as measured by 3T MRI[J]. J Orthop Res, 2010, 28(9): 1149-1154.
[29] Jedrzejczyk T, Mikusek J, Rudnicki P, et al. The infrapatellar adipose body in humans of various age groups[J]. Folia Morphol (Warsz), 1996, 55(1): 51-55.
[30] Conde J, Scotece M, Abella V, et al. Identification of novel adipokines in the joint. Differential expression in healthy and osteoarthritis tissues[J]. PLoS One, 2015, 10(4): e123601.
[31] Conde J, Scotece M, Lopez V, et al. Differential expression of adipokines in infrapatellar fat pad (IPFP) and synovium of osteoarthritis patients and healthy individuals[J]. Ann Rheum Dis, 2014, 73(3): 631-633.
[32] Bastiaansen-Jenniskens YM, Clockaerts S, Feijt C, et al. Infrapatellar fat pad of patients with end-stage osteoarthritis inhibits catabolic mediators in cartilage[J]. Ann Rheum Dis, 2012, 71(2): 288-294.
[33] Gandhi R, Takahashi M, Virtanen C, et al. Microarray analysis of the infrapatellar fat pad in knee osteoarthritis: relationship with joint inflammation[J]. J Rheumatol, 2011, 38(9): 1966-1972.
