泛素系统在结核分枝杆菌与宿主相互作用中的调控机制研究进展①
2017-02-27柴琪瑶刘翠华
柴琪瑶 刘翠华
(中国科学院微生物研究所病原微生物与免疫学重点实验室,北京100101)
·专家述评·
泛素系统在结核分枝杆菌与宿主相互作用中的调控机制研究进展①
柴琪瑶 刘翠华
(中国科学院微生物研究所病原微生物与免疫学重点实验室,北京100101)
结核分枝杆菌(Mycobacterium tuberculosis,Mtb)是一种极其成功的胞内病原菌,可通过多种策略实现免疫逃逸,从而在宿主巨噬细胞中长期存活。在对抗病原菌的防御过程中,泛素系统(Ubiquitin system)在激活宿主炎症免疫反应、细胞自噬、吞噬体成熟和细胞死亡等天然免疫功能及相关信号通路中发挥了重要的调控作用。而另一方面,近年的研究表明Mtb等胞内病原菌可通过分泌效应蛋白(Effector proteins)挟持并利用宿主泛素系统进而抑制宿主的免疫功能,这些病原-宿主互作的界面有望成为抗结核药物研发的新靶点。
泛素系统;结核分枝杆菌;巨噬细胞;天然免疫;信号通路;效应蛋白

柴琪瑶(1992年-),2014年本科毕业于浙江大学植物保护专业,同年推荐免试进入中国科学院微生物研究所病原生物学专业,师从刘翠华教授硕博连读。主要从事结核分枝杆菌和巨噬细胞相互作用的分子机制研究。

刘翠华(1976年-),中国科学院微生物研究所研究员,课题组长,博士生导师。任“中国防痨协会结核病基础专业分会”和“中国免疫学会青年工作委员会”等委员以及Scientific Reports等国际期刊编委。主要从事病原菌感染和宿主免疫防御的分子机制等方面的研究,在结核分枝杆菌逃逸宿主固有免疫机制等方面的研究中取得了一系列成果,在Cell、Nat Immunol、PNAS、J Immunol等期刊发表学术论文40余篇,作为编委或编者参编《泛素介导的蛋白质降解》等专著6部。
由结核分枝杆菌(Mycobacterium tuberculosis,Mtb)引起的结核病(Tuberculosis,TB)是全球十大“致死性疾病”之一。世界卫生组织报告显示,仅2015年全球就有1 040万新增结核病例和140万死亡病例;我国的结核患者数量(约91.8万人)居世界第三位,同时也是耐多药结核病(MDR-TB)和广泛耐药结核病(XDR-TB)病例最多的国家之一[1,2]。由于特殊的潜伏特性以及耐药结核病的出现和泛滥,结核病的防治工作面临巨大的挑战。深入研究Mtb与宿主的相互作用机制、为结核病的快速诊断和新药研发提供新靶点迫在眉睫。
泛素(Ubiquitin,Ub)是真核生物中的一种由76个氨基酸组成的小分子量蛋白,能够通过共价结合的方式连接到底物蛋白分子的赖氨酸残基上而介导靶蛋白的降解[3]。自上世纪70年代被鉴定以来,人们发现泛素不仅是蛋白质降解的信使,而且在细胞增殖和死亡、囊泡运输、转录调控、DNA损伤修复及感染免疫等生理过程中发挥重要的调控作用[4]。本文所讨论的泛素系统包括泛素、泛素化(Ubiqu-itination)或去泛素化(Deubiquitination)修饰相关的酶、泛素结合蛋白或受体等,以及上述蛋白参与的各类泛素修饰过程。这些元素构成的泛素系统在机体天然免疫系统抵御病原菌感染的过程中起到了多种调节作用。Mtb作为一种典型的胞内菌在与宿主长期共存的过程中进化出多种免疫逃逸策略,本文将对泛素系统在结核分枝杆菌与宿主互作过程中的调控机制进行论述,以期为结核感染及致病的分子机制研究提供新的思路。
1 结核分枝杆菌与宿主相互作用的分子机制
Mtb是一种强传染性的胞内病原菌,活动性结核病患者咳出的气溶胶只要携带1~3个活性菌体便可通过呼吸系统感染一个新的健康个体[5]。一般情况下,侵入肺部的非致病分枝杆菌,如耻垢分枝杆菌(M.smegmatis)等将被肺泡巨噬细胞(Alveolar macrophage)识别和吞噬,最终被溶酶体的各类水解酶、活性氮/氧等物质杀灭。然而,致病性的Mtb却能避免被宿主完全清除。在肺表面感染Mtb的巨噬细胞将携带菌体穿过上皮细胞深入感染肺组织,并不断地产生分化和聚集。随后,淋巴细胞(Lymphocyte)和树突状细胞(DC)等多种免疫细胞也会集结到周围,最终在受感染的组织处发展成肉芽肿(Granuloma)[5]。一些学者认为,宿主细胞形成稳固的肉芽肿可在一定程度上限制病原菌的感染和扩散,迫使其进入休止(Quiescence)状态[5,6]。然而,也有研究表明在感染初期Mtb会主动促进肉芽肿的形成,并利用这种特殊结构来侵入新的细胞和组织,帮助其定殖和扩散[5,7]。虽然诸多生物学特性尚未被揭示,但研究者发现只要机体的免疫功能稳健,肉芽肿对宿主的健康并不显示负面影响;只有当机体免疫系统发生衰退或缺陷时,Mtb才会结束漫漫“长征”而伺机活化和大量增殖[7]。换言之,肉芽肿实际上是Mtb与宿主免疫系统之间长期博弈最终达到制衡状态的产物,而Mtb与巨噬细胞的相互作用成为了这场战役中最核心的部分。
进入宿主体内后,Mtb的细胞壁成分如磷脂酰肌醇甘露糖(PIM)、脂质阿拉伯甘露聚糖(LAM)及其他一些外膜分子将被巨噬细胞通过膜表面受体所识别,这些受体有甘露糖受体(MR)、Toll-样受体(TLR)、补体受体(CR)、凝集素受体(DC-SIGN)和免疫球蛋白Fc段受体(Fc receptor)等,并进一步激活下游效应分子而引发宿主免疫防御。以TLR为例,Mtb细胞壁的脂蛋白(Lipoprotein)、脂多糖(Liposaccharide)及CpG DNA可以作为TLR2的配体被识别而激活下游的NF-κB信号通路,并刺激TNF-α的分泌进而活化巨噬细胞,同时激发维生素D受体及维生素D-1-羟化酶基因的表达诱导抗菌肽(Cathelicidin)的释放[8]。被吞噬入胞后,Mtb仍将面临多方面的免疫清除作用。Mtb除了通过内吞/吞噬体途径(Endocytic/phagocytic pathway)被降解外,逃离吞噬体的病原菌还会触发细胞自噬(Autophagy)而被重新捕获和清除。例如,NOD是一类重要的天然免疫胞内识别受体,Mtb的胞壁酰二肽(MDP)受到NOD2的识别后,将激发宿主炎症因子及氮氧化物(NO)的释放来起到杀菌作用,同时活化的NOD2将诱导细胞发生自噬[9]。
然而,作为最古老的传染病之一的Mtb在与宿主长期的协同进化中采取了一系列特殊的免疫逃逸和调控策略来达到对宿主的生境适应(Local adaptation)目的[10]。Mtb有别于其他细菌的一个重要特征是具有复杂的膜结构——由类似革兰氏阴性菌内膜的质膜(Plasma membrane)、富含糖蛋白的周质区(Periplasmic space)以及高度疏水的外膜(Outer membrane)构成,这让体内微生境(Niche)中的Mtb能够有效地隔离有害物质,并使其拥有一套独特的分泌系统[11]。Mtb具有GSP/SecA1、Tat、SecA2及ESAT6四类分泌系统,其中,ESAT6分泌系统又称Ⅶ型分泌系统(T7SS)或Esx分泌系统,能够分泌多种效应蛋白(Effector proteins),成为抵抗宿主免疫系统的利器[11,12]。Mtb能够编码ESX-1~5五种ESAT6分泌系统,以ESX-1为例,该系统由EccB/C/D/E四个膜组分构成跨越细菌质膜的分泌孔道,其中EccC的胞质结构域能够识别EccA并与之形成AAA+ATPase,而EspG扮演分子伴侣(Chaperone)的角色与底物蛋白结合形成稳定的异二聚体,在胞质中EccA/G两个辅助因子的帮助下指导底物蛋白结合到分泌通道复合物上;此外,Mtb的膜蛋白酶MycP还能进一步对ESX-1分泌蛋白进行加工,成熟的效应蛋白随后被递送到菌表面或宿主细胞内发挥多样化的免疫调控作用[12]。目前,对于T7SS系统的结构和机制还所知甚少,ESX系统和分泌底物之间、效应蛋白和细菌毒力之间的微妙联系等都是Mtb致病机制研究的热点。
2 泛素系统调控宿主天然免疫信号通路的机制
2.1 泛素修饰的分子机制 经典的泛素化过程一般需要三种泛素化酶的参与,即通过泛素激活酶(Ubiquitin-activating enzyme,E1)、泛素偶联酶(Ubiquitin-conjugating enzyme,E2)和泛素连接酶(Ubiquitin-ligase enzyme,E3)的协助来活化泛素分子并将其转移到靶蛋白上,形成多聚泛素化(Polyubiquitination)或单泛素化(Monoubiquitionation)修饰[3]。多聚泛素化修饰按泛素之间所连接的赖氨酸位点的不同可以分为K6、K11、K27、K29、K33、K48及K63泛素链;泛素也能够被一类特殊的E3,即线性泛素链组装复合物LUBAC通过N端甲硫氨酸与另一泛素分子的羧基相连,形成线性泛素化(Linear ubiquitination)[13]。然而,最近发现嗜肺军团菌(Legionella pneumophila)分泌的一类SidE家族效应蛋白SdeA具有单-ADP-核糖转移酶模序(Mono-ADP-ribosyltransferase motif),能够通过ADP-核糖基化来活化泛素分子,在无需E1、E2的参与下直接泛素化宿主的多种Rab GTPase,被称为一体化泛素连接酶(All-in-one E3)[14]。此外,蛋白质的泛素化修饰实际上也是一个可逆的过程,与泛素化酶功能相反,真核生物和一些原核生物能够编码一些去泛素化酶(DUBs)来水解并破坏底物蛋白与泛素之间或多聚泛素链内的硫酯键[13]。
泛素除了可以通过共价结合的方式连接到靶蛋白上以外,某些蛋白可直接通过非共价作用结合上单个泛素或泛素链。这类泛素结合蛋白往往存在一个或多个泛素结合域(UBD),虽然这些UBD在三级结构上呈现出多样性,如α-螺旋结构的泛素互作模序(UIM)、锌指(ZnF)结构、pleckstrin同源(PH)折叠结构和泛素偶联酶变体(UEV)等,大部分UBD都是通过识别并结合泛素分子由Leu8、Ile44、His68及Val70组成的疏水氨基酸簇而产生相互作用[15]。综上所述,泛素系统复杂多变的蛋白修饰和互作方式奠定了其在天然免疫信号网络中进行精确调控的分子基础。
2.2 泛素系统对炎症免疫信号通路的调控 模式识别受体(PRR)是一类能够识别病原相关分子模式(PAMP)和损伤相关分子模式(DAMP)的重要免疫受体,包括上述的TLR、NOD样受体(NLR)及RIG-Ⅰ样受体(RLR)等。PRR感应到病原菌入侵后,将通过信号级联网络刺激促炎细胞因子和Ⅰ型干扰素的释放,并刺激NF-κB和MAPK等炎症相关信号通路的活化。例如,巨噬细胞膜表面受体TLR4以病原菌表面的脂多糖(LPS)作为配体,在接头蛋白TIRAP的帮助下集聚转接分子MYD88,后者通过结合IRAK4和IRAK1两类激酶来招募TNF受体相关因子TRAF3/6及凋亡抑制因子cIAP1/2,最终在质膜内侧形成信号级联复合物[16]。其中,具有E3连接酶功能的TRAF6在Ubc13-Uev1a组成的E2复合物的作用下连接K63泛素链而活化,并同时泛素化IRAK1和cIAP1/2[17]。这些K63泛素链在信号网络中起到了脚手架的作用,能够通过结合底物分子(如TAB2/3和NEMO)来进一步募集和激活TAK1和IKK两类激酶复合物;此外,LUBAC介导的NEMO在Lys285与Lys309位点的线性泛素化对NF-κB通路中的信号传递也具有重要意义[18]。TAK1活化后,能够通过激活MAPK激酶MKK4/6/7来刺激JNK及p38信号通路,同时磷酸化激活IKK;而活化的IKK能够磷酸化IκBα并介导其被SCFβTrCP E3连接酶K48泛素化降解,从而解除对NF-κB的抑制,使后者入核调控免疫相关基因的表达[19]。
近年来,炎症小体(Inflammasome)信号通路在调节炎症免疫方面的功能愈来愈受到研究者的重视。炎症小体复合物由受体、凋亡相关斑点样蛋白ASC以及前体caspase-1(pro-caspase-1)所组成。NLR家族如NLRP3、NLRP1、NLRC4及胞质中的DNA感受器AIM2是目前已发现的几类炎症小体受体,感应刺激后,将通过激活caspase-1介导促炎细胞因子IL-1β和IL-18的释放并诱导细胞焦亡(Pyroptosis)[20]。与凋亡(Apoptosis)不同的是,细胞焦亡是炎性caspase(caspase-1和/或caspase-11)活化后发生的炎症性细胞死亡方式,将导致细胞膜破裂出现孔隙,引起细胞溶胀裂解,从而破坏胞内寄生菌定殖的微生境,使其出胞后被其他免疫细胞所吞噬和杀灭。目前,泛素系统对于NLRP3炎症小体的调控机制较为清楚。巨噬细胞在静息状态时,胞内的 E3连接酶SCF的FBXL2催化亚基能够通过K48泛素化修饰NLRP3而介导其降解;当受到病原菌配体LPS刺激时,细胞将诱导提高E3连接酶FBXO3的表达,后者通过泛素化降解FBXL2来维持NLRP3的蛋白水平,起到炎症上调作用[21]。其次,胞质中一类K63去泛素化酶BRCC3能够通过直接去泛素化稳定NLRP3的蛋白水平[21]。另一方面,泛素系统对炎症小体信号通路存在有效的负调控机制,从而避免过度活化的炎症反应导致细胞损伤。例如,巨噬细胞内的去泛素化酶A20可抑制炎症小体复合物中caspase-1对前体IL-1β(pro-IL-1β)的切割活性,同时能够去除pro-IL-1β的K63泛素化而起到负调节作用[22]。另一项对神经递质多巴胺(Dopamine)的研究发现,其能够通过活化细胞内E3连接酶MARCH7来泛素化降解NLRP3而起到抑炎作用[23]。此外,巨噬细胞中存在一类Cullin环E3连接酶抑制子GLMN,对NLRP3和NLRC4两类炎症小体的活化具有抑制功能;然而,这个特性也被肠上皮细胞寄生菌志贺氏杆菌(Shigella)所利用,其分泌的具有E3连接酶功能的IpaH7.8效应蛋白能够通过泛素化介导GLMN的降解,进而解除后者对炎症因子的活化抑制[24]。
2.3 泛素系统对自噬信号通路的调控 自噬发生时,胞质内产生一种称为自噬体(Autophagosome)的双层膜泡结构,能够在自噬相关信号调控下逐渐与溶酶体相融合并降解所包裹的内容物。自噬调控核心由一类自噬相关蛋白(Atg)所组成。哺乳动物细胞中,自噬体的形成主要受到了ULK(酵母Atg1同源)蛋白激酶复合体及VPS34脂激酶复合体的调控。ULK复合体由ULK1/2、FIP200(酵母Atg17同源)、ATG13及ATG101等组成,并受到对细胞营养条件敏感的mTORC1激酶复合体的直接调控[25]。此外,胞质中mTOR激酶的另一种复合体形式mTORC2能够磷酸化Akt激酶,通过活化PI3K-Akt-mTOR信号级联促进mTORC1对自噬的抑制活性,并且该通路中Akt与mTOR的激酶活性还受到了TRAF6介导的K63泛素化的正向调节[26]。脂激酶VPS34通过与p150及Beclin-1形成激酶复合物磷酸化磷脂酰肌醇形成PI3P,PI3P在胞内的膜结构与胞质交界处聚集,为募集自噬体发生相关蛋白提供锚定点,同时能够介导多种效应分子有秩序地调控自噬体的转运和成熟[27]。Beclin-1作为一个重要的转接分子能够结合多种调控蛋白来增强(如UVRAG、ATG14L、Bif-1及AMBRA1等)或抑制(如Bcl-2、TAB2/3及Rubicon等)VPS34的激酶活性,从而起到调节自噬的功能[28]。ULK及VPS34复合体中的多个组分都受到了泛素调控。例如,自噬发生时胞质中的AMBRA1能够募集TRAF6并与ULK1或Beclin-1结合使其K63泛素化,一方面能够稳定及活化ULK复合体,另一方面通过引起Beclin-1寡聚化并阻碍其与Bcl-2结合而起到对VPS34的正调控;相反地,mTORC1则能够通过磷酸化AMBRA1来阻碍其招募TRAF6而起到自噬抑制作用[29]。此外,Beclin-1还受到E3连接酶RNF216的K48泛素化修饰,从而减弱其与VPS34复合物的结合;相反地,去泛素化酶USP19则能够特异性降解Beclin-1的K11泛素链从而维持其稳定性[29]。
近年来,异体自噬(Xenophagy)和线粒体自噬(Mitophagy)这两类选择性自噬机制受到了格外关注。线粒体自噬过程中,PINK1激酶能够在去极化的线粒体外膜聚集,通过磷酸化被泛素化修饰的线粒体蛋白上的泛素分子来招募选择性自噬受体,如OPTN、NDP52等[30]。这些受体由泛素结合域UBA与泛素化蛋白结合,同时通过LIR(LC3 interacting region)模序结合LC3,从而和底物蛋白一起经自噬体转运至溶酶体中发生降解。同时,PINK1也能在线粒体外膜上募集E3连接酶Parkin,后者泛素化多类线粒体蛋白底物(如VDAC、Miro及mitofusin-2等)并介导降解[30]。有趣的是,Parkin也能够介导胞内病原菌的异体自噬。例如,Mtb能通过ESX-1系统在吞噬体上造成孔洞从而释放细菌DNA激活cGAS-STING依赖的胞质DNA感应信号通路,进而诱发Parkin聚集到Mtb吞噬体上并对其标记上K63泛素化修饰引起自噬降解[31]。然而,Parkin针对病原菌或吞噬体膜上的泛素化底物,以及其在菌表面募集和活化的机制目前尚不清楚。除了OPTN及NDP52外,还发现有SQSTM1(p62)和NBR1等多种选择性自噬受体,这些受体在介导选择性自噬时可能并非功能冗余。例如,p62更倾向于介导K63而非K48泛素化标记的底物降解,OPTN则倾向于结合线性泛素链;p62能够与NBR1形成异二聚体协作完成对泛素化底物的自噬介导,同时NBR1也能自身形成低聚化介导过氧化物酶体自噬(Pexophagy);然而p62在介导一些胞内菌如志贺菌、李斯特菌(Listeria)及沙门氏菌(Salmonella)的自噬清除时仅需要NDP52而非NBR1的协助[32]。
2.4 泛素系统对吞噬体成熟信号通路的调控 宿主的巨噬细胞、中性粒细胞以及树突状细胞等专职吞噬细胞对病原菌具有很强的吞噬清除作用。吞噬发生时,首先由Fcγ受体(FcγR)、补体受体3(CR3)等膜表面受体接受病原菌携带的吞噬信号而引起胞内侧的免疫受体模序ITAM的活化,并进一步诱发一系列吞噬相关激酶(如Syk、Cdc42、Rac1/2及PI3K等)的信号级联反应,从而介导肌动蛋白重组和质膜内化形成吞噬体(Phagosome)[33]。随后,吞噬体经历早期和晚期阶段逐渐酸化和成熟,最终与溶酶体融合形成吞噬溶酶体(Phagolysosome)介导菌的降解。
目前发现,哺乳动物细胞存在超过60个成员的Rab家族GTPase,它们是指导吞噬体成熟过程的核心机制。例如,Rab5对介导质膜处发生的初期吞噬体与早期内体(Endosome)的融合具有推进作用。一方面,鸟苷酸交换因子(GEF)Rabex-5在早期吞噬体膜上的募集能够促进Rab5的活化;另一方面活化的Rab5则可通过招募效应分子Rabaptin-5进一步促进Rabex-5的活性,从而形成一个正反馈循环[34]。在这个循环中,泛素调控起到了关键作用。Rabex-5是一类具有E3连接酶活性的泛素结合蛋白,能够通过ZnF及一个反泛素结合模序(IUIM)从胞质中结合到泛素化的吞噬体膜蛋白上行使对Rab5的激活功能;当内体融合完成后, Rabex-5将被单泛素化修饰并转位回到胞质中[34]。此外,内体相关的一类DUBs,如AMSH和UBPY则可通过去泛素化Rabex-5来恢复该循环[35]。伴随早期吞噬体发展为晚期吞噬体的一个重要标识是Rab5向Rab7的转变。Rab7通过募集诸多效应分子来协助吞噬体的进一步成熟,并介导晚期吞噬体与溶酶体的融合。研究发现,Rab7是E3连接酶Parkin的一个底物,被泛素化修饰后的Rab7能够增强其在膜上的稳定性及招募效应分子的能力,从而促进吞噬体成熟[36]。
在胞质中,有一类具有封闭膜结构并包裹大量腔内小泡的多泡小体(MVB)也受到了吞噬体的识别和降解,并影响吞噬体的发展和病原菌的清除能力。例如,由ESCRT-0/Ⅰ/Ⅱ/Ⅲ组成的转运相关内体分选复合物(ESCRT)是指导MVB形成的主要分子机制,而Mtb的Esx-3效应蛋白EsxH和EsxG能够形成复合物靶向破坏ESCRT的功能,从而抑制吞噬体的成熟[37]。实验表明,泛素系统在调控ESCRT复合物介导的MVB生物合成过程具有重要作用。ESCRT-0/Ⅰ/Ⅱ复合物能够通过募集多种含不同类型UBD的泛素结合蛋白,如STAM1/2(UIM)、Hrs(UIM)、Tsg101(Uev)及EAP45(GLUE)等捕获和聚集泛素化货物于分隔小泡中,随后ESCRT-Ⅲ启动膜泡出芽和分裂,促使包被货物小泡的MVB形成[38]。
2.5 泛素系统对细胞死亡信号通路的调控 如前所述,泛素系统能够通过炎症及自噬信号通路来影响细胞焦亡和自噬性细胞死亡;而凋亡和坏死(Necrosis)作为天然免疫信号通路中一类重要机制同样受到了泛素系统的精密调控。细胞凋亡根据诱发的起始因素的不同可分为外源性凋亡途径(Extrinsic pathway)和内源性凋亡途径(Intrinsic pathway)。外源途径诱导凋亡时,最先通过细胞表面一类接受凋亡信号(如TNF-α、FasL及TRAIL等)的死亡受体(DR),如TNFR1、DR3、DR4、DR5及Fas等介导激活。例如,TNF-α与TNFR1形成复合体激活后可以通过受体结合激酶RIPK1依赖或非依赖的两种途径传递凋亡信号,且都受到了泛素系统的调控。其中,RIPK1依赖的凋亡信号通路被认为可能发生在线粒体介导的内源性凋亡途径的下游。因为当接受凋亡信号后,线粒体外膜通透性改变而促使一类凋亡相关蛋白Smac被释放到胞质中,随后与凋亡抑制蛋白cIAP结合并使后者发生自降解而丧失对多种caspase的抑制作用;同时去泛素化酶CYLD、USP7或USP2a通过介导RIPK1的去泛素化使其脱离TNFR1受体复合物,转而在胞质中招募含有死亡结构域(DD)的接头蛋白FADD及caspase-8,最终形成另一种死亡诱导信号复合物(DISC)并介导凋亡[39,40]。相应的研究表明,通过线性泛素链被招募到TNFR1复合物中的另一种去泛素化酶A20则能够通过自身的ZnF与泛素分子相互作用来起到稳定泛素链的作用[41]。RIPK1非依赖的凋亡信号通路则是由胞质中一类通过竞争性结合FADD而抑制caspase-8活性的蛋白c-FLIP所决定,且受到了泛素系统的调控。c-FLIP包括c-FLIPL、c-FLIPS及c-FLIPR三种亚型,例如在Mtb引起的活性氧(ROS)依赖的巨噬细胞凋亡途径中,涉及到ASK1、p38及c-Abl三类激酶的激活并引发c-FLIPS的酪氨酸/丝氨酸磷酸化,促进后者与E3连接酶c-Cbl的结合及泛素化降解,从而解除对caspase-8的抑制作用[42]。
在感染初期,Mtb能够通过抑制细胞凋亡来促进自身的胞内存活,而感染晚期的Mtb却通过诱导细胞自噬来逃离衰竭的细胞并扩散感染[43]。与凋亡所不同的是,细胞发生坏死时将出现质膜破裂和内容物的外溢,同时引起炎症免疫反应。近年来,人们发现非caspase或细胞色素C介导的死亡实际上也受到了复杂的信号网络调控,并把这类细胞死亡方式归入程序性细胞坏死(Necroptosis)的范畴。程序性坏死的调控核心是RIPK3及MLKL激酶。在TNFR1介导的凋亡信号通路中,若caspase-8活性降低或胞质中RIPK3和MLKL蛋白水平较高时,RIPK3将通过RHIM结构域与RIPK1结合并自磷酸化,活化后的RIPK3通过招募特异性底物MLKL作为坏死执行者激活坏死信号通路[44]。如前所述,泛素系统能够通过对RIPK1的修饰起到对胞质中caspase-8活化水平的调节,从而决定细胞凋亡或程序性坏死的发展方向。近年来还发现胞内调控p53和p21泛素化降解而起到凋亡调控作用的E3连接酶MKRN1也能通过泛素化FADD来抑制caspase-8的活性,同时影响坏死小体(Necrosome)的形成[45]。另一方面,RIPK3和MLKL本身也存在泛素化修饰的现象,例如鼠源RIPK3在K5位置能够被A20泛素化从而促进其与RIPK1形成复合物,然而MLKL的泛素化修饰机制目前尚不清楚[46]。
3 结核分枝杆菌利用泛素系统逃避免疫清除的机制
效应蛋白是Mtb在宿主体内长期存活并逃逸免疫清除的最大助力(图1)。目前所发现的Mtb效应蛋白的功能至少有6个方面:①促进Mtb的侵染入胞,如Erp、FbpA、Mce3家族等细胞壁蛋白能够被分泌到环境中帮助Mtb黏附和感染宿主[11,47];②具有抗氧化/氮化应激作用,如SodA、KatG、AhpC及TpX等酶类能够抵抗胞内的活性氧/氮介质(ROI/RNI)对Mtb的胁迫[11,48];③参与细胞凋亡的调控,近年来鉴定出MPT64、Rv3354及PtpA等多种效应蛋白具有凋亡抑制功能,例如MPT64能够增强宿主巨噬细胞Bcl-2蛋白家族的表达水平,而PtpA则能够去磷酸化增强GSK3α的抗凋亡活性[49];④抑制吞噬体的成熟,如Mtb分泌的一类核苷二磷酸激酶Ndk能够抑制Rab5和Rab7与各自的效应分子EEA1或RILP的结合[50];又如PtpA及Mtb分泌的另一种磷酸酶SapM,前者能够去磷酸化宿主的膜泡转运蛋白VPS33B,同时结合溶酶体膜上的V-ATPase的H亚基来阻止吞噬体与吞噬体膜相融合[51];后者则能去磷酸化吞噬体膜上的PI3P阻碍吞噬体发展[52];⑤抑制炎症信号通路的活化,例如,PknE能够抑制人THP-1巨噬细胞的TNF-α和IL-6炎症因子的分泌水平[11,53];而Mtb分泌的金属蛋白酶Zmp1则能够抑制caspase-1的活性,进而抑制pro-IL-1β的切割活化,并阻碍炎症小体的形成[54]。最近发现Mtb膜效应蛋白Mce3E在感染过程中会进入巨噬细胞内靶向结合MAPK信号通路的关键激酶Erk1/2,并滞留活化的Erk1/2在内质网中而阻碍其入核调控基因转录,同时抑制TNF-α和IL-6的表达水平[55];⑥抑制细胞自噬,例如Mtb效应蛋白EIS能够增强组蛋白H3的乙酰化水平来上调巨噬细胞的IL-10的基因表达水平,通过活化Akt/mTOR/p70S6K信号通路来抑制细胞自噬[56]。最近还鉴定出分枝杆菌效应蛋白PE_PGRS47也具有自噬抑制功能,该蛋白缺失的Mtb感染巨噬细胞将显著增多胞质中LC3的聚集,且p62自噬受体的降解明显增强[57]。
利用泛素系统逃避宿主的免疫清除是近年来发现的一种Mtb的重要免疫逃逸机制,该机制有赖于Mtb效应蛋白PtpA等的调控功能。研究发现:PtpA存在一个全新的类泛素结合模序UIML,能够通过非共价结合到单泛素分子的疏水氨基酸簇上来增强自身磷酸酶活性,并靶向宿主MAPK信号通路中两类关键激酶JNK和p38,或吞噬体成熟相关蛋白VPS33B起到去磷酸化抑制的作用;同时还可通过竞争性结合TAB3的NZF锌指结构抑制后者与K63泛素化的结合进而阻断NF-κB信号通路,并降低TNF-α及IL-1β的表达水平[58]。近年来,还鉴定出Mtb的一类蛋白激酶Rv3354具有保护Mtb分泌蛋白不被宿主泛素化降解的功能。Rv3354能够靶向结合宿主的一类蛋白质降解调控复合物——COP9信号小体(CSN)的JAMM结构域并抑制其对泛素样蛋白Nedd8的切割活性,而Nedd8的切割是CNS调控宿主CRL家族E3连接酶复合物活化的关键[59]。此外,在Mtb感染过程中,诸多泛素相关基因出现了明显的上调(如mkrn1、cops5等)或下调(zfp91、ndfip2、ube2f、rnft1、psmb6和psmd13等)现象,这暗示泛素系统在Mtb-宿主相互作用过程中具有极为重要的调控功能[60]。
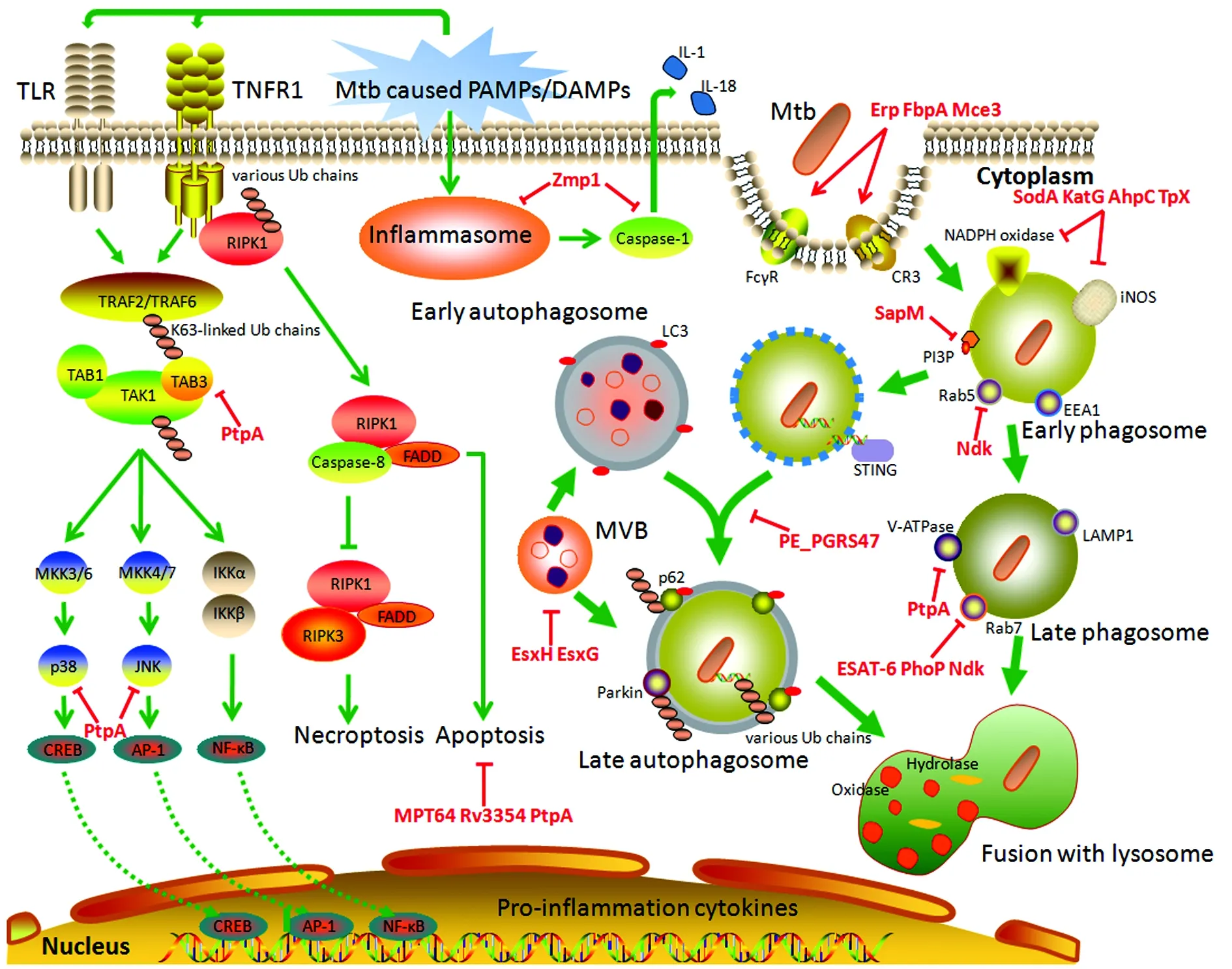
图1 结核分枝杆菌效应蛋白的免疫抑制功能Fig.1 Immunosuppressive functions of M.tuberculosis effector proteinsNote: Mtb effector proteins play a pivotal role in orchestrating host innate immune signaling pathway.Erp,FbpA and Mce3 are essential for maintaining Mtb virulence during the infection[11,47].The antioxidant enzymes SodA,KatG,AhpC and TpX confer Mtb resistance to host oxidative/nitrosative stresses[11,48].SapM,Ndk and PtpA arrest phagosome by targeting PI3P,Rab5/7 and V-ATPase,respectively[50-52].The Rab7 recruitment is also undermined by ESAT-6 and PhoP[64].PE_PGRS47 reduces LC3 puncta and p62 degradation,whereas EsxH and EsxG impair MVB formation to inhibit autophagy[57].PtpA competitively interacts with TAB3 to prevent the binding of K63 Ub chains and directly dephosphorylates phos-p38 and phos-JNK to inhibit both NF-κB and MAPK pathway[58].MPT64,Rv3354 and PtpA have different apoptotic regulatory functions when delivered into cytoplasm[49].ZMP1 acts as a Zn2+ metalloprotease with the ability to restrain the host inflammasome development and suppress the activity of caspase-1[54].
有趣的是,宿主并不甘于被动地接受Mtb对泛素系统的挟持。最近的一项研究发现,宿主的一类E3连接酶Trim27能够通过RING结构域结合PtpA,对PtpA等Mtb效应蛋白引起的巨噬细胞天然免疫抑制功能产生对抗作用,进而限制分枝杆菌的胞内存活,这类具有抵御病原菌作用的宿主蛋白被称为限制因子(Restriction factors)[61]。事实上,宿主泛素系统威胁Mtb胞内存活的另一张王牌是依赖泛素系统的选择性自噬机制。除了Parkin介导的Mtb泛素化,最近还发现宿主范醌蛋白1(UBQLN1)也能够通过Mtb的ESAT-6效应蛋白EsxA与菌表面发生相互作用,进而促进泛素、p62及LC3在Mtb表面聚集并介导Parkin非依赖性的自噬降解[62]。此外,宿主对Mtb的选择性自噬也可能通过TLR2活化的DNA损伤相关自噬调节子DRAM1介导而聚集LC3,该过程无需泛素及p62等受体的参与[32,63]。然而,巨噬细胞胞质中的Mtb强毒株H37Rv和弱毒株H37Ra虽然都能被自噬体捕获,但前者明显能够抑制自噬体的进一步成熟及与溶酶体融合。研究发现,Mtb强毒株能够通过阻止自噬体招募Rab7来抑制其与内体形成中间囊泡(Amphisome)的成熟过程,而这种能力有赖于ESAT-6分泌系统及其调节因子PhoP的协助[64]。此外,Mtb通过调控宿主miRNA影响自噬相关基因的表达是新近发现的一种免疫逃逸机制。Mtb感染过程中将诱导增强巨噬细胞miR-33及其过客链(passenger strand)miR-33*的表达水平,而miR-33和miR-33*的上调表达将负向调控一系列自噬相关蛋白(如ATG5、ATG12、LC3B及LAMP1等)编码基因的表达水平,改变宿主的自噬调控程序[65]。
4 展望
综上所述,越来越多的研究表明:泛素系统的免疫调控功能对宿主抵御病原菌感染具有重要作用,但这一特点也能被病原菌所利用。目前已在沙门氏菌、李斯特菌、志贺菌、嗜肺军团菌、耶尔森氏菌(Yersinia)、肠出血性大肠杆菌(EHEC)及肠致病性大肠杆菌(EPEC)等多种病原菌中鉴定出一系列具有真核细胞E3连接酶或DUBs活性的效应蛋白,这些病原菌可通过模拟宿主泛素系统的功能进而扰乱宿主防御机制并达到免疫逃逸的目的[66]。然而,有关这些病原菌效应蛋白在病原-宿主互作过程中的调控机制方面的研究仅是冰山一角,很多未知的问题有待进一步探寻。例如,这些效应蛋白在原核生物中是否普遍存在或具有同源性蛋白?在感染过程中它们究竟受到何种时间或空间依赖性的精细调控?其作用是否具有宿主和细胞特异性?宿主又有哪些限制因子用来规避或降解这些病原菌效应蛋白?针对以上一系列问题,建立合适的病原-宿主感染模型,并应用多学科交叉的手段从病原菌感染免疫、蛋白分子生化修饰、细胞信号通路级联调控等角度深入研究病原菌效应蛋白的调控功能和机制,将有望为感染性疾病的防控提供新策略以及为抗感染药物研发提供新靶点。
[1] World Health Organization.Global tuberculosis report 2016[R].World Health Organization,Geneva,Switherland 2016,WHO/HTM/TB/2016.13.
[2] Pang Y,Dong HY,Tan YJ,etal.Rapid diagnosis of MDR and XDR tuberculosis with the MeltPro TB assay in China[J].Sci Rep,2016,6:25330.
[3] Morreale FE,Walden H.Types of ubiquitin ligases[J].Cell,2016,165(1):248.
[4] Hu R,Hochstrasser M.Recent progress in ubiquitin and ubiquitin-like protein (Ubl) signaling[J].Cell Res,2016,26(4):389-390.
[5] Cambier CJ,Falkow S,Ramakrishnan L.Host evasion and exploitation schemes of Mycobacterium tuberculosis[J].Cell,2014,159(7):1497-1509.
[6] McDaniel MM,Krishna N,Handagama WG,etal.Quantifying limits on replication,death,and quiescence of mycobacterium tuberculosis in mice[J].Front Microbiol,2016,7:862.
[7] Martin CJ,Carey AF,Fortune SM.A bug's life in the granuloma[J].Semin Immunopathol,2016,38(2):213-220.
[8] Yang CS,Shin DM,Kim KH,etal.NADPH oxidase 2 interaction with TLR2 is required for efficient innate immune responses to mycobacteria via cathelicidin expression[J].J Immunol,2009,182(6):3696-3705.
[9] Chauhan S,Mandell MA,Deretic V.Mechanism of action of the tuberculosis and Crohn disease risk factor IRGM in autophagy[J].Autophagy,2016,12(2):429-431.
[10] Brites D,Gagneux S.Co-evolution of Mycobacterium tuberculosis and Homo sapiens[J].Immunol Rev,2015,264:6-24.
[11] Forrellad MA,Klepp LI,Gioffre A,etal.Virulence factors of the Mycobacterium tuberculosis complex[J].Virulence,2013,4(1):3-66.
[12] Groschel MI,Sayes F,Simeone R,etal.ESX secretion systems:mycobacterial evolution to counter host immunity[J].Nat Rev Microbiol,2016,14(11):677-691.
[13] Clague MJ,Heride C,Urbe S.The demographics of the ubiquitin system[J].Trends Cell Biol,2015,25(7):417-426.
[14] Qiu J,Sheedlo MJ,Yu K,etal.Ubiquitination independent of E1 and E2 enzymes by bacterial effectors[J].Nature,2016,533(7601):120-124.
[15] Husnjak K,Dikic I.Ubiquitin-binding proteins:decoders of ubiquitin-mediated cellular functions[J].Annu Rev Biochem,2012,81:291-322.
[16] He A,Ji R,Shao J,etal.TLR4-MyD88-TRAF6-TAK1 complex-mediated NF-kappaB activation contribute to the anti-inflammatory effect of V8 in LPS-induced human cervical cancer siHa cells[J].Inflammation,2016,39(1):172-181.
[17] Chen ZJ.Ubiquitination in signaling to and activation of IKK[J].Immunol Rev,2012,246:95-106.
[18] Tokunaga F,Sakata S,Saeki Y,etal.Involvement of linear polyubiquitylation of NEMO in NF-kappaB activation[J].Nat Cell Biol,2009,11(2):123-132.
[19] Häcker H,Karin M.Regulation and Function of IKK and IKK-related Kinases[J].Sci STKE,2006,2006(357):re13.
[20] Lechtenberg BC,Riedl SJ.Rosetta stone of NLR innate immunity[J].Trends Biochem Sci,2016,41(1):6-8.
[21] Bednash JS,Mallampalli RK.Regulation of inflammasomes by ubiquitination[J].Cell Mol Immunol,2016,13(6):722-728.
[22] Duong BH,Onizawa M,Oses-Prieto JA,etal.A20 restricts ubiquitination of pro-interleukin-1beta protein complexes and suppresses NLRP3 inflammasome activity[J].Immunity,2015,42(1):55-67.
[23] Yan Y,Jiang W,Liu L,etal.Dopamine controls systemic inflammation through inhibition of NLRP3 inflammasome[J].Cell,2015,160(1-2):62-73.
[24] Ashida H,Sasakawa C.Shigella IpaH family effectors as a versatile model for studying pathogenic bacteria[J].Front Cell Infect Microbiol,2015,5:100.
[25] Ktistakis NT,Tooze SA.Digesting the expanding mechanisms of autophagy[J].Trends Cell Biol,2016,26(8):624-635.
[26] Kim YC,Guan KL.mTOR:a pharmacologic target for autophagy regulation[J].J Clin Invest,2015,125(1):25-32.
[27] Vicinanza M,Rubinsztein DC.Mirror image phosphoinositides regulate autophagy[J].Mol Cell Oncol,2016,3(2):e1019974.
[28] Fu LL,Cheng Y,Liu B.Beclin-1:autophagic regulator and therapeutic target in cancer[J].Int J Biochem Cell Biol,2013,45(5):921-924.
[29] Antonioli M,Di Rienzo M,Piacentini M,etal.Emerging mechanisms in initiating and terminating autophagy[J].Trends Biochem Sci,2017,42(1):28-41.
[30] Lazarou M,Sliter DA,Kane LA,etal.The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy[J].Nature,2015,524(7565):309-314.
[31] Shibutani ST,Saitoh T,Nowag H,etal.Autophagy and autophagy-related proteins in the immune system[J].Nat Immunol,2015,16(10):1014-1024.
[32] Khaminets A,Behl C,Dikic I.Ubiquitin-dependent and independent signals in selective autophagy[J].Trends Cell Biol,2016,26(1):6-16.
[33] Jaumouille V,Grinstein S.Molecular mechanisms of phagosome formation[J].Microbiol Spectr,2016,4(3):1-18.
[34] Kalin S,Hirschmann DT,Buser DP,etal.Rabaptin5 is recruited to endosomes by Rab4 and Rabex5 to regulate endosome maturation[J].J Cell Sci,2015,128(22):4126-4137.
[35] Hologne M,Cantrelle FX,Riviere G,etal.NMR reveals the interplay among the AMSH SH3 binding motif,STAM2,and Lys63-Linked diubiquitin[J].J Mol Biol,2016,428(22):4544-4558.
[36] Song P,Trajkovic K,Tsunemi T,etal.Parkin modulates endosomal organization and function of the endo-lysosomal pathway[J].J Neurosci,2016,36(8):2425-2437.
[37] Tinaztepe E,Wei JR,Raynowska J,etal.Role of metal-dependent regulation of ESX-3 secretion in intracellular survival of mycobacterium tuberculosis[J].Infect Immun,2016,84(8):2255-2263.
[38] Christ L,Raiborg C,Wenzel EM,etal.Cellular functions and molecular mechanisms of the ESCRT membrane-scission machinery[J].Trends Biochem Sci,2017,42(1):42-56.
[39] Dillon CP,Oberst A,Weinlich R,etal.Survival function of the FADD-CASPASE-8-cFLIPL complex[J].Cell Rep,2012,1(5):401-407.
[40] Lopez-Castejon G,Edelmann MJ.Deubiquitinases:novel therap-eutic targets in immune surveillance?[J].Mediators Inflamm,2016,2016:3481371.
[41] Draber P,Kupka S,Reichert M,etal.LUBAC-recruited CYLD and A20 regulate gene activation and cell death by exerting opposing effects on linear ubiquitin in signaling complexes[J].Cell Rep,2015,13(10):2258-2272.
[42] Kundu M,Pathak SK,Kumawat K,etal.A TNF- and c-Cbl-dependent FLIP(S)-degradation pathway and its function in Mycobacterium tuberculosis-induced macrophage apoptosis[J].Nat Immunol,2009,10(8):918-926.
[43] Pesu M.New insights into the host cell necrosis in tuberculosis[J].Virulence,2016,7(1):1-2.
[44] Remijsen Q,Goossens V,Grootjans S,etal.Depletion of RIPK3 or MLKL blocks TNF-driven necroptosis and switches towards a delayed RIPK1 kinase-dependent apoptosis[J].Cell Death Dis,2014,5:e1004.
[45] Lee EW,Kim JH,Ahn YH,etal.Ubiquitination and degradation of the FADD adaptor protein regulate death receptor-mediated apoptosis and necroptosis[J].Nat Commun,2012,3:978.
[46] Onizawa M,Oshima S,Schulze-Topphoff U,etal.The ubiquitin-modifying enzyme A20 restricts ubiquitination of the kinase RIPK3 and protects cells from necroptosis[J].Nat Immunol,2015,16(6):618-627.
[47] Meena LS,Rajni.Survival mechanisms of pathogenic Mycobact-erium tuberculosis H37Rv[J].FEBS J,2010,277(11):2416-2427.
[48] Hu Y,Coates AR.Acute and persistent Mycobacterium tuberculosis infections depend on the thiol peroxidase TpX[J].PLoS One,2009,4(4):e5150.
[49] Liu M,Li W,Xiang X,etal.Mycobacterium tuberculosis effectors interfering host apoptosis signaling[J].Apoptosis,2015,20(7):883-891.
[50] Sun J,Wang X,Lau A,etal.Mycobacterial nucleoside diphosphate kinase blocks phagosome maturation in murine RAW 264.7 macrophages[J].PLoS One,2010,5(1):e8769.
[51] Wong D,Bach H,Sun J,etal.Mycobacterium tuberculosis protein tyrosine phosphatase (PtpA) excludes host vacuolar-H+-ATPase to inhibit phagosome acidification[J].Proc Natl Acad Sci U S A,2011,108(48):19371-19376.
[52] Puri RV,Reddy PV,Tyagi AK.Secreted acid phosphatase (SapM) of Mycobacterium tuberculosis is indispensable for arresting phagosomal maturation and growth of the pathogen in guinea pig tissues[J].PLoS One,2013,8(7):e70514.
[53] Kumar D,Narayanan S.pknE,a serine/threonine kinase of Mycobacterium tuberculosis modulates multiple apoptotic paradigms[J].Infect Genet Evol,2012,12(4):737-747.
[54] Master SS,Rampini SK,Davis AS,etal.Mycobacterium tuberculosis prevents inflammasome activation[J].Cell Host Microbe,2008,3(4):224-232.
[55] Li J,Chai QY,Zhang Y,etal.Mycobacterium tuberculosis Mce3E suppresses host innate immune responses by targeting ERK1/2 signaling[J].J Immunol,2015,194(8):3756-3767.
[56] Duan L,Yi M,Chen J,etal.Mycobacterium tuberculosis EIS gene inhibits macrophage autophagy through up-regulation of IL-10 by increasing the acetylation of histone H3[J].Biochem Biophys Res Commun,2016,473(4):1229-1234.
[57] Saini NK,Baena A,Ng TW,etal.Suppression of autophagy and antigen presentation by Mycobacterium tuberculosis PE_PGRS47[J].Nat Microbiol,2016,1(9):16133.
[58] Wang J,Li BX,Ge PP,etal.Mycobacterium tuberculosis suppresses innate immunity by coopting the host ubiquitin system[J].Nat Immunol,2015,16(3):237-245.
[59] Danelishvili L,Babrak L,Rose SJ,etal.Mycobacterium tuberculosis alters the metalloprotease activity of the COP9 signalosome[J].MBio,2014,5(4):e01278-e01314.
[60] Meenu S,Thiagarajan S,Ramalingam S,etal.Modulation of host ubiquitin system genes in human endometrial cell line infected with Mycobacterium tuberculosis[J].Med Microbiol Immunol,2016,205(2):163-171.
[61] Wang J,Teng JL,Zhao D,etal.The ubiquitin ligase TRIM27 functions as a host restriction factor antagonized by Mycobacter-ium tuberculosis PtpA during mycobacterial infection[J].Sci Rep,2016,6:34827.
[62] Sakowski ET,Koster S,Portal CC,etal.Ubiquilin 1 promotes IFN-gamma-induced xenophagy of Mycobacterium tuberculosis[J].PLoS Pathog,2015,11(7):e1005076.
[63] Meijer AH,van der Vaart M.DRAM1 promotes the targeting of mycobacteria to selective autophagy[J].Autophagy,2014,10(12):2389-2391.
[64] Chandra P,Ghanwat S,Matta SK,etal.Mycobacterium tuberculosis inhibits RAB7 recruitment to selectively modulate autophagy flux in macrophages[J].Sci Rep,2015,5:16320.
[65] Ouimet M,Koster S,Sakowski E,etal.Mycobacterium tuberculosis induces the miR-33 locus to reprogram autophagy and host lipid metabolism[J].Nat Immunol,2016,17(6):677-686.
[66] Li J,Chai QY,Liu CH.The ubiquitin system:a critical regulator of innate immunity and pathogen-host interactions[J].Cell Mol Immunol,2016,13(5):560-576.
[收稿2016-11-18]
(编辑 倪 鹏)
Regulatory mechanisms of ubiquitin system in Mycobacterium tuberculosis-host interactions
CHAIQi-Yao,LIUCui-Hua.
CASKeyLaboratoryofPathogenicMicrobiologyandImmunology,InstituteofMicrobiology,ChineseAcademyofSciences,Beijing100101,China
Mycobacterium tuberculosis (Mtb) is an extremely successful intracellular bacterial pathogen that engages multiple strategies to escape host immune surveillance,so as to survive within host macrophages for long period.Upon the invasion of pathogenic bacteria,the host ubiquitin system plays a critical role in activating the host innate immune responses and associated signaling pathways such as inflammatory and immune responses,autophagy,phagosome maturation and cell death,etc.On the other hand,recent studies have demonstrated that intracellular pathogenic bacteria such as Mtb can secrete a variety of effector proteins into host cells to hijack or co-opt the ubiquitin system to suppress host immune responses.Those pathogen-host interacting interfaces could provide potential novel targets for the development of anti-tuberculosis drugs.
Ubiquitin system;Mycobacterium tuberculosis;Macrophage;Innate immunity;Signaling pathway;Effector proteins
10.3969/j.issn.1000-484X.2017.02.001
R392
A
1000-484X(2017)02-0161-09
①本文受国家自然科学基金(No. 81371769, 81571954)、国家973基础科学项目(2014CB744400)和中国科学院“青年创新促进会优秀会员”项目(Y12A027BB2)资助。
