新型吡咯并三嗪酮类PARP-1抑制剂的设计、合成及生物活性
2017-02-24刘志雄王小龙李福卫于海涛
刘志雄, 王小龙, 薛 鑫, 李福卫, 于海涛
(南京中医药大学 药物合成技术研究中心,江苏 南京 210023)
·研究论文·
新型吡咯并三嗪酮类PARP-1抑制剂的设计、合成及生物活性
刘志雄, 王小龙, 薛 鑫, 李福卫, 于海涛*
(南京中医药大学 药物合成技术研究中心,江苏 南京 210023)
分别以5-溴-2-氟苯甲腈(1a)和3-溴苯甲腈(1b)为原料,经Sonogashira偶联,脱三甲基硅基保护基,三分子偶联及水解等5步反应制得中间体2-氟-5-[(4-氧代-3,4-二氢吡咯[1,2-d][1,2,4]三嗪-1-基)甲基]苯甲酸(6a)和3-[(4-氧代-3,4-二氢吡咯[1,2-d][1,2,4]三嗪-1-基)甲基]苯甲酸(6b)。环烷基甲酸经酰氯化,缩合和脱Boc保护基3步反应制得环烷基哌嗪-1-基甲酮(7a~7c)。 6a与NCS(1 eq.)反应制得5-[(6-氯-4氧代-3,4二氢吡咯[1,2-d][1,2,4]三嗪-1-基)甲基]-2氟-苯甲酸(6c); 6a与NCS(2 eq.)反应制得5-[(6,7-二氯-4氧代-3,4二氢吡咯[1,2-d][1,2,4]三嗪-1-基)甲基]-2氟-苯甲酸(6d)。 6a~6d, 6a~6c分别与7a~7c和1-(2-嘧啶基)哌嗪在TBTU(缩合剂),DIPEA(碱)的作用下合成了13个新型吡咯并三嗪酮类PARP-1抑制剂(8a~8m),其结构经1H NMR和MS(ESI)表征。采用Alarm blue法研究了8a~8m对肿瘤细胞MDA-MB-436的抑制活性(IC50)。结果表明:8f, 8g, 8i和8j对MDA-MB-436有较强的抑制活性(IC50=30.5~69.3 nmol·L-1)。
苯甲腈; 吡咯并三嗪酮; PARP-1抑制剂; 合成; 生物活性
聚腺苷二磷酸核糖聚合酶(PARP)是一种家族酶,它们催化ADP核糖单元从烟酰胺腺嘌呤二核苷酸(DAD+)转移至各种蛋白底物上。该家族酶包括17个成员,其中PARP-1研究最为广泛。PARP-1的主要功能为作为DNA修复因子,其效果在碱基切除修复过程中尤为显著[1-6]。
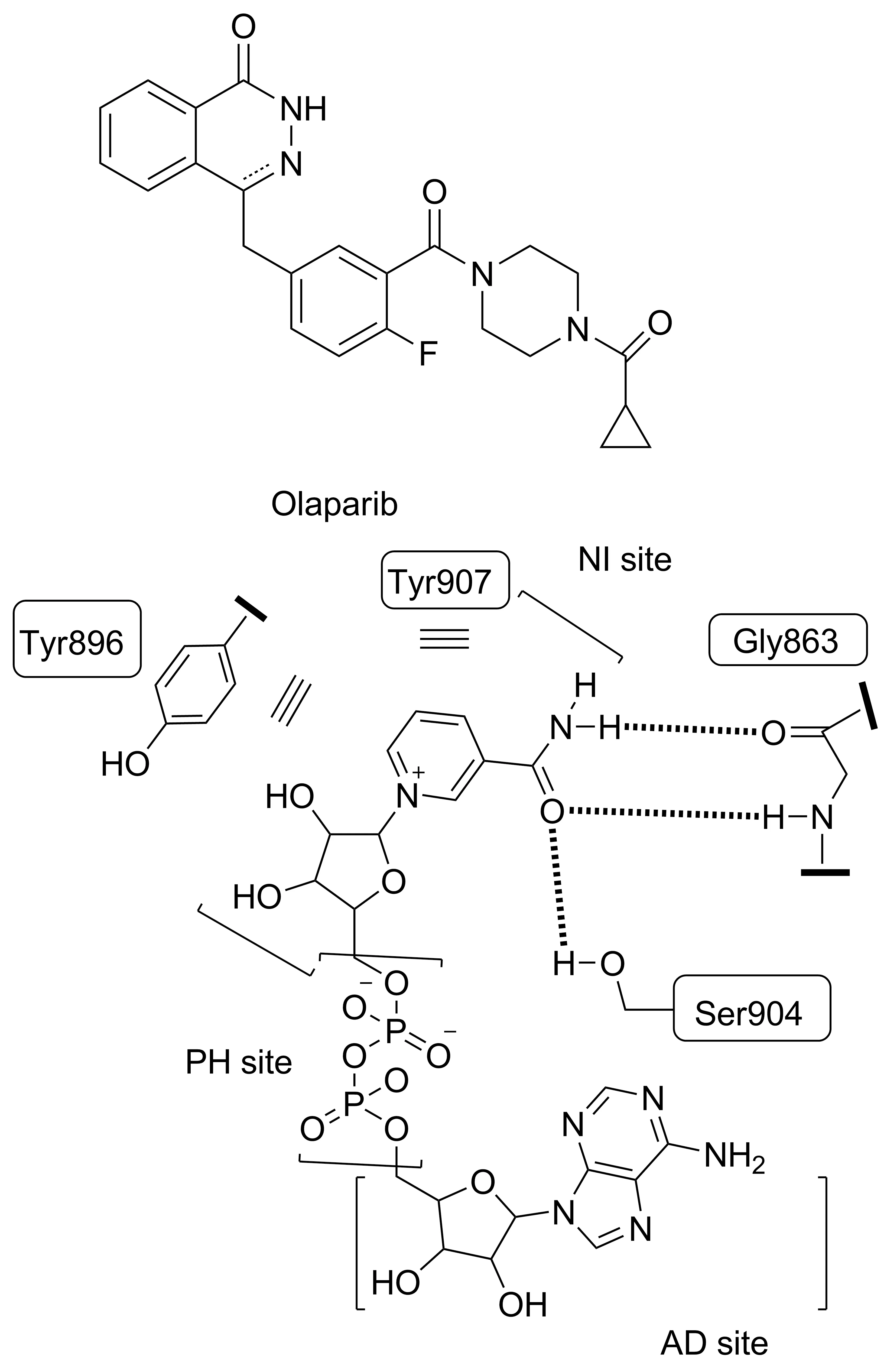
Chart 1
2014年12月,FDA批准了全球首种PARP-1抑制剂——奥拉帕尼(Olaparib),用于治疗BRCA基因突变的卵巢癌患者。研究表明,PARP-1的C端催化区的天然底物为NAD+,根据催化区域位置和功能的不同,可以分为3个结合位点[7]:烟酰胺-核糖结合位点(NI site)、磷酸结合位点(PH site)和腺苷-核糖结合位点(AD site)。现有的PARP-1抑制剂大多通过与NAD+竞争性结合PARP-1的NI site而产生活性[8]。NAD+与NI site的结合模式为:(1)吡啶环与PARP-1蛋白的Tyr896和Tyr907产生π-π堆积作用。(2)甲酰胺基团与PARP-1蛋白的Gly863和Ser904形成3个氢键(Chart 1)[9]。
本文通过分析Olaparib的分子结构特点和NAD+与PARP-1的作用机制,设计并合成了未见文献报导的新型吡咯并三嗪酮类PARP-1抑制剂(8a~8m)并研究了其生物活性。(1)将苯环改变成吡咯环,增加芳香环的富电性,考察对这种π-π堆积作用力的影响。减小A环的空间体积,使抑制剂继续深入PARP-1的V字形口袋,考察该改变对抑制剂与PARP-1蛋白的氢键结合力的影响。(2)保留哌嗪环为碱性末端以调节水溶性,并以PARP-1抑制剂结构中常见的环丙基羰基、2-嘧啶基及环戊基羰基连接哌嗪环(Chart 2)。

Chart 2
以5-溴-2-氟苯甲腈(1a)和3-溴苯甲腈(1b)为原料,经Sonogashira偶联,脱三甲基硅基保护基,三分子偶联及水解等5步反应制得中间体2-氟-5-[(4-氧代-3,4-二氢吡咯[1,2-d][1,2,4]三嗪-1-基)甲基]苯甲酸(6a)和3-[(4-氧代-3,4-二氢吡咯[1,2-d][1,2,4]三嗪-1-基)甲基]苯甲酸(6b)。环烷基甲酸经酰氯化,缩合和脱Boc保护基3步反应制得环烷基哌嗪-1-基甲酮(7a~7c)。 6a与NCS(1 eq.)反应制得5-[(6-氯-4氧代-3,4二氢吡咯[1,2-d][1,2,4]三嗪-1-基)甲基]-2氟-苯甲酸(6c); 6a与NCS(2 eq.)反应制得5-[(6,7-二氯-4氧代-3,4二氢吡咯[1,2-d][1,2,4]三嗪-1-基)甲基]-2氟-苯甲酸(6d)。 6a~6d, 6a~6c分别与7a~7c和1-(2-嘧啶基)哌嗪在TBTU(缩合剂),DIPEA(碱)的作用下合成了13个新型吡咯并三嗪酮类PARP-1抑制剂(8a~8m),其结构经1H NMR和MS(ESI)表征。采用Alarm blue法研究了8a~8m对肿瘤细胞MDA-MB-436的抑制活性(IC50)。

Scheme 1
1 实验部分
1.1 仪器与试剂
X-5型显微熔点仪;Bruker Ascend 400 MHz型核磁共振仪(DMSO-d6为溶剂,TMS为内标);Agilent 1100型液质联用仪;Bio-Tek Synergy HT型多标记微孔板检测仪;Invitrogen C10281 Countess型自动细胞计数器。
人乳腺癌细胞(MDA-MB-436),美国模式菌种收集中心;Alarm Blue试剂,Sigma公司;其余所用试剂均为分析纯或化学纯。
1.2 合成
(1) 2a和2b的合成(以2a为例)
在反应瓶中依次加入1a 12.0 g(60.00 mmol),碘化亚铜0.576 g(3.0 mmol), PdCl2(PPh3)22.1 g(2.9 mmol),三乙胺25.2 mL(181.5 mmol),三甲基硅乙炔8.52 mL(60.28 mmol)和THF 200 mL,氮气保护,搅拌下回流(60 ℃)反应18 h(TLC监测)。冷却至室温,加入饱和氯化铵水溶液400 mL,分液,水相用乙酸乙酯(3×100 mL)萃取,合并有机相,用饱和NaCl溶液(300 mL)洗涤,无水硫酸钠干燥,减压蒸干,残余物经硅胶柱层析[洗脱剂:A=V(EA) ∶V(PE)=1 ∶20]纯化得棕色油状液体2-氟-5-三甲基硅烷乙炔基苯甲腈(2a)9.96 g,产率76.6%。
用类似的方法合成棕色油状液体3-(三甲基硅基乙炔基)苯甲腈2b,收率80%[10]。
(2) 3a与3b的合成(以3a为例)
在反应瓶中依次加入四正丁基氟化铵35.9 g(137.30 mmol)和THF 200 mL,于室温缓慢滴加至2a 9.96 g(45.83 mmol)的THF(400 mL)溶液中,反应完全(TLC监测)。加入饱和氯化铵水溶液400 mL,分液,水相用乙酸乙酯(3×100 mL)萃取,合并有机相,用饱和NaCl溶液(300 mL)洗涤,无水硫酸钠干燥,减压旋干,残余物经硅胶柱层析[A=V(EA) ∶V(PE)=3 ∶1]纯化得黄色固体5-乙炔基-2-氟苯甲腈(3a)5.86 g,产率88.1%。
用类似的方法合成黄色固体3b,产率83.0%[10]。
(3) 4a和4b的合成(以4a为例)
在反应瓶中依次加入对甲苯磺酰叠氮6.0 g(30.42 mmol),氯化亚铜0.3 g(3.04 mmol), 4a 5.3 g(36.51 mmol)和氯仿60 mL,氮气保护,于室温搅拌10 min;依次加入三乙胺5.10 mL(36.51 mmol)和吡咯6.30 mL(91.27 mmol),于45 ℃反应4 h(TLC监测)。加入饱和氯化铵水溶液100 mL,分液,水相用二氯甲烷(3×50 mL)萃取,合并有机相,用饱和NaCl溶液(300 mL)洗涤,无水硫酸钠干燥,减压旋干得灰色固体N-[2-(3-氰基-4-氟苯基)-1-(1H-吡咯-2-基)亚乙基]-4-甲基苯磺酰胺(4a)7.95 g,产率68.5%; MS(ESI)m/z: 382.1{[M+H]+}。
用类似的方法合成灰色固体4b,产率70.0%; MS(ESI)m/z:364.1{[M+H]+}[11]。
(4) 5a和5b的合成(以5a为例)
在反应瓶中依次加入4a 7.0 g(18.35 mmol),肼基甲酸乙酯2.8 g(26.89 mmol)和叔丁醇50 mL,氮气保护,于室温搅拌10 min;加入叔丁醇钾6.0 g(53.47 mmol),回流(80 ℃)反应12 h(TLC监测)。冷却至室温,加入水100 mL,用1 mol·L-1盐酸溶液调至pH≈7,用乙酸乙酯(3×100 mL)萃取,合并有机相,用饱和NaCl溶液(300 mL)洗涤,无水硫酸钠干燥,减压旋干,残余物经硅胶柱层析(洗脱剂:A=1 ∶10)纯化得黄色粉末2-氟-5-(4-氧代-3,4-二氢吡咯并[1,2-d][1,2,4]三嗪-1-基-甲基)苯甲腈(5a)4.21 g,产率86.3%;1H NMRδ: 12.18(s, 1H), 7.93(dd,J=6.2 Hz, 2.2 Hz, 1H), 7.81~7.70(m, 2H), 7.46(t,J=9.1 Hz, 1H), 6.93(dd,J=3.7 Hz, 1.2 Hz, 1H), 6.83~6.76(m, 1H), 4.15(s, 2H); MS(ESI)m/z: 269.0{[M+H]+}。
用类似的方法合成黄色粉末5b,产率79.6%;1H NMRδ: 12.19(s, 1H), 7.86(s, 1H), 7.72(dd,J=17.0 Hz, 4.7 Hz, 1H), 7.52(t,J=7.8 Hz, 1H), 6.93(d,J=3.4 Hz, 1H), 6.80(t,J=3.3 Hz, 1H), 4.17(s, 2H); MS(ESI)m/z: 251.1{[M+H]+}。
(6) 6a与6b的合成(以6a为例)
在反应瓶中依次加入5a 1.8 g(6.71 mmol),氢氧化钠1.4 g(35.00 mmol)和水30 mL,于室温搅拌10 min,回流(90 ℃)反应8 h(TLC监测)。冷却至室温。滴加1 mol·L-1盐酸溶液调至pH酸性,析出白色固体,过滤,滤饼用混合溶液(A=1 ∶5)洗涤,真空干燥得白色固体6a 1.8 g,产率93.3%;1H NMRδ: 13.20(s, 1H), 11.84(s, 1H), 7.79(dd,J=7.1 Hz, 2.3 Hz, 1H), 7.62~7.48(m, 1H), 7.37~6.99(m, 3H), 6.31~6.13(m, 1H), 4.14 (s, 2H); MS(ESI)m/z: 288.0{[M+H]+}。
用类似的方法合成白色固体6b;1H NMRδ: 13.10(s, 1H), 11.74(s, 1H), 7.75(s, 1H), 7.54~7.24(m, 4H), 6.91(s, 1H), 6.80(s, 1H), 4.14(s, 1H); MS(ESI)m/z: 270.0{[M+H]+}。
(7) 6c和6d的合成(以6c为例)
将NCS 0.496 g(3.71 mmol, 1 eq.)溶于二氯甲烷30 mL中,氮气保护下,于-5 ℃依次加入四氯化锆86.6 mg(0.371 mmol)和6a 1.0 g(3.71 mmol),加毕,缓慢升至室温,反应完全(TCL监测)。加水50 mL,析出固体,过滤,滤饼用混合溶液(A=1 ∶5)洗涤,真空干燥得淡黄色固体6c 0.89 g,产率80.0%;1H NMRδ: 12.04(s, 1H), 7.69(dd,J=7.0 Hz, 2.2 Hz, 1H), 7.44~7.36(m, 1H), 7.06(dd,J=10.4 Hz, 8.5 Hz, 1H), 6.83(d,J=4.0 Hz, 1H), 6.72(d,J=4.0 Hz, 1H), 4.00(s, 2H); MS(ESI)m/z: 322.0{[M+H]+}。
仅改变NCS为2 eq.,用类似的方法合成黄色固体6d, 产率86%;1H NMRδ: 12.20(s, 1H), 7.75(dd,J=7.1 Hz, 2.3 Hz, 1H), 7.55~7.48(m, 1H), 7.24(dd,J=10.7 Hz, 8.6 Hz, 1H), 6.99(s, 1H), 4.25(s, 2H); MS(ESI)m/z: 356.0{[M+H]+}[12]。
(8) 7a~7c的合成(以7c为例)
将环戊酸1.0 g(8.76 mmol)溶于氯化亚砜(10 mL)中,回流2 h。旋干氯化亚砜,残余物用二氯甲烷10 mL溶解;缓慢滴加N-Boc哌嗪1.7 g(9.13 mmol)的二氯甲烷(10 mL)溶液,滴毕,搅拌下于室温反应2 h。旋干二氯甲烷,残余物真空干燥得白色固体4-环戊基基甲酰基哌嗪-1-甲酸叔丁酯2.3 g,产率93%。
将4-环戊基-甲酰基哌嗪-1-甲酸叔丁酯2.3 g(8.14 mmol)溶于无水甲醇(20 mL)中,剧烈搅拌下通入干燥HCl气体(1 h);析出大量白色固体,停止通气,于室温反应1 h。旋干反应液,残余物加入碳酸钠溶液20 mL,用二氯甲烷(3×30 mL)萃取,合并有机相,依次用大量饱和碳酸钠溶液和氯化钠饱和溶液洗涤,无水Na2SO4干燥,旋干,残余物真空干燥得无色油状液体环戊基-哌嗪-1-基甲酮(7c)1.41 g,产率95%;1H NMRδ: 3.57(t,J=4.0 Hz, 2H), 3.47(t,J=4.0 Hz, 2H), 2.84~2.87(m, 5H), 2.21(s, 1H), 1.65~1.77(m, 6H), 1.48~1.58(m, 2H); MS(ESI)m/z: 183.1{[M+H]+}。
用类似的方法合成无色油状液体环丁基-哌嗪-1-基甲酮(7b), 产率91%;1H NMRδ: 3.48~3.16(m, 6H), 2.70~2.55(m, 5H), 2.22~1.63(m, 5H); MS(ESI)m/z: 169.1{[M+H]+}。
无色油状液体环丙基-哌嗪-1-基甲酮(7a),产率92%;1H NMRδ: 3.65(t,J=4.0 Hz, 2H), 3.44(t,J=6.0 Hz, 2H), 3.32(m, 3H), 3.29(m, 2H), 1.97(s, 1H), 0.73(dt,J=9.5 Hz, 5.3 Hz, 4H); MS(ESI)m/z: 155.1{[M+H]+}。
(9) 8a~8m的合成(以8b为例)
在反应瓶中依次加入6b 2.0 g(7.43 mmol), 7c 1.4 g(7.68 mmol),苯并三氮唑-N,N,N′,N′-四甲基脲四氟硼酸酯(TBTU)3.6 g(11.21 mmol),N,N-二异丙基乙胺2.9 g(22.44 mmol)和DMF 20 mL,搅拌下于室温反应3 h。加入水100 mL,用1 mol·L-1盐酸调至pH=7,析出大量固体,继续滴加盐酸至无固体析出。过滤,滤饼依次用大量水和少量乙醇洗涤,真空干燥得白色固体1-[3-(4-环戊基羰基-1-哌嗪-1-甲酰基)苄基]-3H-吡咯并[1,2-d][1,2,4]三嗪-4-酮(8b)3.0 g,产率93.0%[13]。
用类似的方法合成白色固体8a, 8c~8m。
8a: 产率91.0%, m.p.131.2~134.3 ℃;1H NMRδ: 7.29(d,J=7.3 Hz, 1H), 6.91(s, 1H), 6.80(s, 1H), 4.14(s, 2H), 3.90~3.00(m, 8H), 0.84(m, 1H), 0.75~0.72(m, 4H); MS(ESI)m/z: 406.1{[M+H]+}。
8b: 产率92.1%, m.p.142.2~145.4 ℃;1H NMRδ: 12.21(s, 1H), 7.75(d,J=7.6 Hz,1H), 7.45~7.37(m, 3H), 7.27(d,J=7.4 Hz,1H), 6.91(d,J=2.6 Hz, 1H), 6.80(t,J=3.2 Hz, 1H), 4.13(s, 2H), 3.54~3.27(m, 8H), 1.65~1.53(m, 8H), 0.88~0.82(m, 1H); MS(ESI)m/z: 434.2{[M+H]+}。
8c: 产率92.1%, m.p.126.1~131.0 ℃;1H NMRδ: 12.20(s, 1H), 7.74(d,J=1.5 Hz, 1H), 7.42~7.58(m, 2H), 7.25(t,J=9.0 Hz, 1H), 6.91(d,J=2.8 Hz, 1H), 6.80(d,J=2.9 Hz, 1H), 4.13(s, 2H), 3.19~3.76(m, 8H), 1.99~2.04(m, 1H), 0.72~0.75(m, 4H); MS(ESI)m/z: 424.1{[M+H]+}。
8d: 产率93.2%, m.p.139.1~142.2 ℃;1H NMRδ: 12.20(s, 1H), 7.75(d,J=1.6 Hz, 1H), 7.43(dd,J=32.8 Hz, 13.2 Hz, 2H), 7.25(t,J=9.0 Hz, 1H), 6.91(d,J=2.6 Hz, 1H), 6.8(s, 1H), 4.11(s, 2H), 3.54~3.27(m, 8H), 1.65~1.53(m, 8H), 0.88~0.82(m, 1H); MS(ESI)m/z: 452.2{[M+H]+}。
8e: 产率92.1%, m.p.109.5~114.8 ℃;1H NMRδ: 12.06(s, 1H), 7.46(ddd,J=12.5 Hz, 7.6 Hz, 5.2 Hz, 1H), 7.41(d,J=5.4 Hz, 1H), 7.25(s, 1H), 6.90(d,J=4.0 Hz, 1H), 6.80(d,J=4.0 Hz, 1H), 4.06(s, 2H), 3.82~3.09(m, 8H), 1.33~0.61(m,5H); MS(ESI)m/z: 458.1{[M+H]+}。
8f: 产率93.2%, m.p.115.1~117.4 ℃;1H NMRδ: 12.06(s, 1H), 7.46(dd,J=7.1 Hz, 4.5 Hz, 1H), 7.39(d,J=6.3 Hz, 1H), 7.24(t,J=9.0 Hz, 1H), 6.89(t,J=3.9 Hz, 1H), 6.79(d,J=4.0 Hz, 1H), 4.05(s, 2H), 3.63~3.10(m, 8H), 2.23~1.58(m, 7H); MS(ESI)m/z: 472.1{[M+H]+}。
8g: 产率93.6%, m.p.116.6~119.4 ℃;1H NMRδ: 12.06(s, 1H), 7.47(s, 1H), 7.40(d,J=5.8 Hz, 1H), 7.24(t,J=8.9 Hz, 1H), 6.89(s, 1H), 6.79(d,J=4.0 Hz, 1H), 4.05(s, 2H), 3.55~3.18(m, 8H), 1.84~1.24(m, 9H); MS(ESI)m/z: 486.1{[M+H]+}。
8h: 产率92.1%, m.p.119.2~121.4 ℃;1H NMRδ: 12.20(s, 1H), 7.39(d,J=5.9 Hz, 1H), 7.33~7.29(m, 1H), 7.24(t,J=9.0 Hz, 1H), 6.98(s, 1H), 4.23(s, 2H), 3.84~3.12(m, 8H), 0.95~0.61 (m, 5H); MS(ESI)m/z: 492.1{[M+H]+}。
8i: 产率94.2%, m.p.123.1~125.3 ℃;1H NMRδ: 12.22(s, 1H), 7.37(s, 1H), 7.29(d,J=6.4 Hz, 1H), 7.23(t,J=9.0 Hz, 1H), 6.99(s, 1H), 4.22(s, 2H), 3.65~3.06(m, 8H), 2.22~1.61(m, 7H); MS(ESI)m/z: 506.1{[M+H]+}。
8j: 产率91.3%, m.p.126.1~129.3 ℃;1H NMRδ: 12.23(s, 1H), 7.38(s, 1H), 7.30(dd,J= 6.4 Hz, 2.0 Hz, 1H), 7.24(t,J=8.0 Hz, 1H), 6.99(s, 1H), 4.23(s, 2H), 3.68~2.84(m, 8H), 1.82~1.12(m, 9H); MS(ESI)m/z: 520.1{[M+H]+}。
8k: 产率91.4%, m.p.198.5~220.3 ℃;1H NMRδ: 12.21(s, 1H), 8.39(d,J=4.7 Hz, 2H), 7.75(dd,J=2.8 Hz, 1.2 Hz, 1H), 7.48~7.38(m, 3H), 7.30(d,J=7.5 Hz, 1H), 6.92(dd,J=3.7 Hz, 1.2 Hz, 1H), 6.80(t,J=3.0 Hz, 1H), 6.68(t,J=4.7 Hz, 1H), 4.14(s, 2H), 3.82~3.27(m, 8H); MS(ESI)m/z: 416.2{[M+H]+}。
8l: 产率93.1%, m.p.220.2~223.1 ℃;1H NMRδ: 9.44(s, 1H), 8.35(d,J=4.7 Hz, 2H), 7.77(d,J=1.6 Hz, 1H), 7.40~7.28(m, 2H), 7.08(t,J=8.7 Hz, 1H), 6.73(t,J=3.3 Hz,1H), 6.57(t,J=4.7 Hz, 1H), 6.66(d,J=2.7 Hz, 1H), 4.08(s, 2H), 3.84~3.40(m, 8H); MS(ESI)m/z: 434.2{[M+H]+}。
8m: 产率94.1%, m.p.225~228.4 ℃;1H NMRδ: 12.06(s, 1H), 8.39(d,J=4.7 Hz, 2H), 7.50~7.45(m, 1H), 7.42(dd,J=6.5 Hz, 2.2 Hz, 1H), 7.25(t,J=9.0 Hz, 1H), 6.91(d,J=4.1 Hz, 1H), 6.79(d,J=4.0 Hz, 1H), 6.68(t,J=4.7 Hz, 1H), 4.06(s, 2H), 3.87~3.79(m, 2H), 3.75~3.65(m, 4H), 3.31~3.24 (m, 2H); MS(ESI)m/z: 468.1{[M+H]+}。
1.3 抗肿瘤活性测试
(1) 细胞培养
MDA-MB-436解冻后,置于含10%胎牛血清、100 U·mL-1青霉素和100 μg·mL-1链霉素的MEM培养基中,放入二氧化碳培养箱,在37 ℃, 5%CO2条件下培养。铺板前,应使细胞传两代以上。
(2) 受试化合物配制
以DMSO为空白溶媒,AZD2281为阳性对照。分别称取适量AZD2281和受试化合物,加入适量DMSO,涡旋振荡使其完全溶解,分装后放入冰箱(-80 ℃)保存。
(3) IC50的测定
检查每板细胞密度,保证细胞处于对数生长期。用0.25%胰蛋白酶消化细胞后制成单细胞悬液,计数。细胞悬液离心5 min(2 000~3 000 r·min-1),取上清液,用少量新配培养液润洗,沉淀,合并上清液,稀释,调整细胞浓度为1×104个/mL。使用12-通道移液枪将细胞悬液按100 mL/孔加入到96孔细胞培养板中,余下每孔各加PBS 200 μL。将96孔板置于孵箱内培养过夜。每孔加入含系列浓度受试化合物的完全培养液或对照的完全培养液100 μL。将板置于孵箱内培养72 h。去除每孔中的培养液,每孔加入含有相应药物的新鲜培养液200 μL,继续培养72 h。将1 mmol·L-1Alarm blue溶液22 μL加至96孔板各孔中后,培养7 h。振荡10 s,测试波长530 nm和590 nm处各孔的荧光强度,计算IC50[14-17]。
2 结果与讨论
2.1 合成
合成5c和5d的关键是控制5a与NCS的当量比,反应温度和反应时间。当5a与NCS的当量比为1 ∶1,反应温度控制在0 ℃以下,反应时间控制在0.5 h内时,大部分产物为一氯代物5c,极少量产物为二氯代物5d,产率约80%。当5a与NCS的当量比为1 ∶2,反应温度控制在0 ℃以下,反应时间控制在0.5 h内时,大部分产物为二氯代物5d,少部分产物为一氯代物和三氯代物,产率约86%。
2.2 生物活性和初步构效关系
表1为目标化合物对MDA-MB-436肿瘤细胞的抑制活性。

表1 8a~8m对MDA-MB-436的抑制活性Table 1 Inhibition activities of 8a~8m against MDA-MB-436
从表1可以看出,8f, 8g, 8i, 8j的抑制活性较好(IC50<100 nmol·L-1),但均比对照药Olaparib差。这说明增加芳香环的富电性不能增强π-π堆积作用力。8a与8c, 8b与8d活性相当,说明结构中引入F不能显著增强抑制剂活性,这可能是因为H和F体积接近所致。8e和8h的抑制活性明显优于8c, 8j和8g明显优于8d, 8m优于8l,说明增大A环的体积可以提高化合物活性,其可能原因为:环体积增大使抑制剂与PARP-1蛋白结合更紧密,增大了相互之间的氢键作用力。8f与8g活性明显优于8e, 8i与8j活性明显优于8h,说明末端取代优先选择体积较大的基团,这可能是因为大基团有助于增强抑制剂与PARP-1蛋白之间的氢键作用力。
以5-溴-2-氟苯甲腈(3-溴苯甲腈)为原料,合成了13个新型的吡咯并三嗪酮类PARP-1抑制剂(8a~8m)。采用Alarm blue法研究了8a~8m对MDA-MB-436肿瘤细胞的抑制活性。结果表明:8f, 8g, 8i和8j的抑制活性较好(IC50<100 nmol·L-1)。初步构效关系表明:(1)增加芳香环富电性不能增加抑制活性;(2)结构中引入氟原子不能显著增加化合物活性;(3)吡咯环上有一氯取代和二氯取代的化合物活性明显优于无取代物;(4)末端取代时,环丁基与环戊基取代化合物活性明显优于环丙基。
[1] Ferraris. Evolution of poly(ADP-ribose)polymerase-1(PARP-1)inhibitors[J].Med Chem,2010,53:4561-4584.
[2] Ding J, Miao Z H, Meng L H,etal. Emerging cancer therapeutic opportunities target DNA-repair systems[J].Trends Pharmacol,2006,27:338-344.
[3] He J X, Yang C H, Miao Z H. Poly(ADP-ribose) polymerase inhibitors as promising cancer therapeutics[J].Acta Pharmacol Sin,2010,31:1172-1180.
[4] Vyas S, Chang P. New PARP targets for cancer therapy[J].Nat Rev Cancer,2014,14:502-509.
[5] Morales J, Li L, Fattah F J,etal. Review of poly(ADP-ribose)polymerase(PARP)mechanisms of action and rationale for targeting in cancer and other diseases[J].Crit Rev Eukaryol Gene Expr,2014,24:15-28.
[6] Benafif S, Hall M. An update on PARP inhibitors for the treatment of cancer[J].Onco Targets Ther,2015,8:519-528.
[7] Ruf A, Gilbert M, Schulz G E. Inhibitor and NAD+binding to poly(ADP-ribose)polymerase as derived from crystal structures and homology modeling[J].Biochemistry,1998,37(11):3893-3900.
[8] Ruf A, Gilbert M, Schulz G E. Structure of the catalytic fragment of poly(ADP-ribose)polymerase from chicken[J].Proc Natl Acad Sci,1996,93(15):7481-7485.
[9] 李福卫. 新型PARP-1抑制剂的设计、合成以及抗肿瘤活性研究[D].南京:南京中医药大学,2016.
[10] Akritopoulou-Zanze I, Wakefield B, D-Gasiecki A. Design,synthesis and biological evaluation of novel 5-substituted indazoles as potent and selective kinase inhibitors employing [2+3]cycloadditions[J].Bioorganic & Medicinal Chemistry Letters,2011,21(5):1476-1479.
[11] Cho S H W, Chang S. Room temperature copper-catalyzed 2-functionalization of pyrrole rings by a three-component coupling reaction[J].Angewandte Chemie,International Edition,2008,47(15):2836-2839.
[12] Zhang Y H, Shi B T, Kazutaka M. Lewis acid-catalyzed highly selective halogenation of aromatic compounds[J].Quick View Other Sources Synlett,2005,(18):2837-2842.
[13] Menear K. 4-[3-4-Cyclopropanecarbonylpiperazine-1-carbonyl)-4-fluorobenzyl]-2H-phthalazin-1-one:A novel bioavalilable inhibitor of poly(ADP-ribose)polymerase[J].Journal of Medicinal Chemistry,2008,51(20):6581-6591.
[14] 杨阳,刘宝瑞,钱晓萍. AlamarBlue法用于体外培养细胞活性检测的方法研究[J].现代肿瘤医学,2006,14(1):006-008.
[15] 高丕尧,龙军先,甘嘉亮,等. CCK-8法与Alamar Blue法对RKO细胞株增殖活力测试条件的对比分析[J].华西药学杂志,2015,30(1):039-041.
[16] 范能全,彭兰. Alamarblue法测定细胞毒性[J].华西药学杂志,2007,22(4):472-474.
[17] 郑梅琴,陆宇,王彬,等. Alamar blue法检测药物细胞内抗结核活性的研究[J].2011,36(6):468-473.
Design, Synthesis and Biological Activities of Novel PARP-1 Inhibitors with Pyrazolotriazinone Skeleton
LIU Zhi-xiong, WANG Xiao-long, XUE Xin, LI Fu-wei, YU Hai-tao*
(Research Center of Pharmaceutical Synthesis Technology, Nanjing University of Chinese Medicine, Nanjing 210023, China)
The intermediates, 2-fluoro-5-[(4-oxo-3,4-dihydropyrrolo[1,2-d][1,2,4]triazin-1-yl)methyl]benzoic acid(6a) and 3-[(4-oxo-3,4-dihydropyrrolo[1,2-d][1,2,4]triazin-1-yl)methyl]benzoic acid(6b) were obtained by a five-step reaction of Sonogashira coupling, deprotection, triplet coupling, hydrolysis, etc, using 5-bromo-2-fluorobenzonitrile(1a) and 3-bromobenzonitrile(1b) as the materials, respectively. Cycloalkylpiperazin-1-ylmethanone(7a~7c) were prepared by a three-step reaction of chlorination, condensation and deprotection from cycloalkylcarboxylic acid. 5-(6-Chloro-4-oxo-3,4-dihydropyrrolo[1,2-d][1,2,4] triazin-1-yl)-2-fluoro-benzoic acid(6c) and 5-[(6,7-dichloro-4-oxo-3,4-dihydropyrrolo [1,2-d][1,2,4]-yl) methyl]-2-fluoro-benzoic acid(6d) were obtained by the reaction of 6a with NCS(1 eq. or 2 eq.). Thirteen novel PARP-1 inhibitors(8a~8m) with pyrazolotriazinone skeleton were synthesized by the condensation reaction of 6a~6d, 6a~6c with 7a~7c or 1-(2-pyrimidyl) piperazine, using TBTU as condensation agent and DIPEA as base. The structures were characterized by1H NMR and MS(ESI). The inhibition activities(IC50) of 8a~8m against tumor cells MDA-MB-436 were investigated by the Alarm blue method. The results indicated that 8f, 8g, 8i and 8j exhibited well activities with IC50of 30.5~69.3 nmol·L-1.
benzonitrile; pyrazolotriazinone; PARP-1 inhibitor; synthesis; biological activity
2016-11-02
国家自然科学基金资助项目(81302650); 江苏高校优势学科建设工程资助项目
刘志雄(1991-),男,汉族,江西吉安人,硕士研究生,主要从事药物合成的研究。 E-mail: m15195758151@163.com
于海涛,博士,讲师, E-mail: njubilly@njucm.edu.cn
O626; O621.3
A
10.15952/j.cnki.cjsc.1005-1511.2017.02.16280
