藏药甘露酥油丸质量标准研究
2017-02-21次仁旺姆拥宗卓玛
次仁旺姆++拥宗卓玛

【摘要】目的:建立藏药甘露酥油丸的质量标准。方法:运用薄层色谱法对样品进行鉴别;采用高效液相色谱法测定没食子酸的含量:Waters Xselect C18 (250 mm × 46 mm,5 μm)柱,流动相:甲醇-02%磷酸溶液(3∶97),检测波长270 nm;柱温25℃;流速为10 mL/min,进样量10μL。结果:诃子、毛诃子、余甘子的薄层鉴别色谱图斑点清晰,分离度好,阴性对照无干扰;没食子酸浓度在05149~10298μg范围内与其峰面积积分值呈良好的线性关系(R2=09999),精密度、重复性、稳定性实验的RSD<2%,平均加样回收率为9714%。结论:本研究制定甘露酥油丸的质量标准,为该品种的质量控制提供了参考依据。
【关键词】甘露酥油丸;高效液相色谱法;薄层色谱;没食子酸
【中图分类号】R917【文献标志码】 A【文章编号】1007-8517(2017)01-0009-03
Abstract:
Keywords:
甘露酥油丸系藏族验方,收载于1995年版《中华人民共和国卫生部药品标准》藏药第一册。甘露酥油丸具有滋补强身、延年益寿等作用,临床用于气血亏虚,精液衰损,心悸失眠,老年虚弱,肢体僵直,经络不利,虚损不足之症[1]。在原标准中只有性状,检测方法有限,因此有必要对甘露酥油丸标准方法进行研究,以建立全面完善的质量标准。
1仪器与材料
11仪器Spectra system液相色谱仪(Waters公司)。Mettler XP205型(精密度为十万分之一)和Mettler XP504(精密度为万分之一)电子天平。
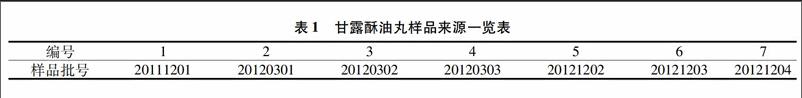
12材料没食子酸对照品(批号:110831-200803,纯度为901%,中国生物制品检定研究院);甘露酥油丸(西藏昌都光宇利民药业有限责任公司)此次样品收集到7批,见表1。 HPLC用色谱纯乙腈和甲醇为Tieda产品,水为纯净水,其它试剂为分析纯。
2薄层色谱鉴别
21诃子薄层色谱鉴别[2]取本品50g,加无水乙醇30mL,加热回流30min,滤过,滤液蒸干,残渣用甲醇5mL使溶解,通过中性氧化铝柱(100~200目,5g,内径为15cm)用稀乙醇50mL洗脱,收集洗脱液,蒸干,残渣用水5mL溶解后通过C18(300mg)固相萃取小柱,用30%甲醇10mL洗脱,弃去30%甲醇液,再用甲醇10mL洗脱,收集洗脱液,蒸干,残渣加甲醇1mL使溶解,作为供试品溶液。取诃子对照药材,按供试品溶液制备方法制备,即得对照药材溶液。取缺诃子的阴性样品,按供试品溶液制备方法制备,即得阴性样品溶液。 按照中国药典2015版通则0502)[3]试验,分别吸取供试品溶液10μL和对照药材溶液5μL,分别点于同一高效硅胶G薄层板上,以甲苯-冰醋酸-水(12 ∶[KG-*2]10 ∶[KG-*2]1)[JP+1]为展开剂,展开,取出,晾干。喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰。供试品色谱中,在与对照药材色谱相应位置上显相同的蓝色斑点。见图1。[FL)]
[LM]
[FL(2K2]
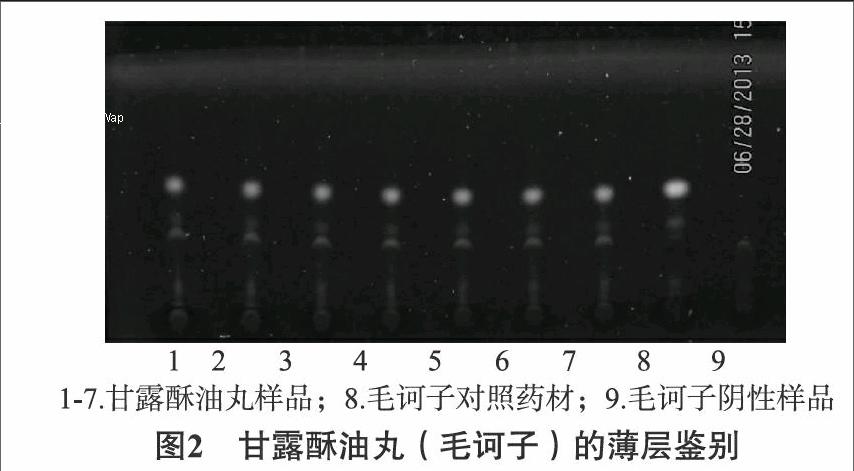
22毛诃子薄层色谱鉴别取本品90g,加热水20mL搅拌使溶解,离心,取上清液,加乙醇30mL,充分搅匀,静置2h,滤过,滤液蒸至约10mL,搅匀,通过聚酰胺柱(30~60目,5g,内径为15cm,干法),加水50mL洗脱,收集洗脱液,置水浴浓缩至约20mL,用醋酸乙酯提取2次,每次20mL,合并醋酸乙酯液,加水20mL,摇匀,放置分层,分取上层液蒸干,残渣加甲醇2mL溶解,作为供试品溶液。另取毛诃子对照药材10g,加热水20mL煮沸,保持微沸30min,滤过,滤液置水浴浓缩至约10mL,用醋酸乙酯提取2次,每次15mL,合并醋酸乙酯液,蒸干,残渣加甲醇2mL溶解,即得对照药材溶液。缺毛诃子的阴性样品,按供试品溶液制备方法制备,即得阴性样品溶液。按照中国药典2015版通则0502[3]试验,吸取上述两种溶液各2μL,分别点于同一硅胶G薄层板上,以甲苯-二氯甲烷-醋酸酯-甲醇(8∶5∶3∶2)为展开剂,展开,取出,晾干。在紫外光灯(365nm)下检视。供试品色谱中,在与对照药材色谱相应位置上显相同的荧光斑点。见图2。
23[JP2]余甘子薄层色谱鉴别取本品90g,加热水20mL搅拌使溶解,离心,取上清液,加乙醇30mL,充分搅匀,静置2h,滤过,滤液蒸至约10mL,搅匀,通过聚酰胺柱(30~60目,5g,内径为15cm,干法),加水50mL洗脱,收集洗脱液,置水浴浓缩至约20mL,用醋酸乙酯提取2次,每次20mL,合并醋酸乙酯液,加水20mL,摇匀,放置分层,分取上层液蒸干,残渣加甲醇2mL溶解,作为供试品溶液。另取余甘子对照药材10g,加热水20mL煮沸,保持微沸30min,滤过,滤液置水浴浓缩至约10mL,用醋酸乙酯提取2次,每次15mL,合并醋酸乙酯液,蒸干,残渣加甲醇2mL溶解,作为对照药材溶液。取缺余甘子的阴性样品,按供试品溶液制备方法制备,即得阴性样品溶液。按照中国药典2015版通则0502[3]试验,吸取上述两种溶液各2μL,分别点于同一硅胶G薄层板上,以二氯甲烷-醋酸乙酯-甲醇-甲酸(9∶9∶3∶02)为展开剂,展开,取出,晾干。喷以10%硫酸乙醇溶液,在105℃加热至浅黄色斑点出现,置紫外光灯(365nm)下检视。供试品色谱中,在与对照药材色谱相应位置上显相同的荧光斑点。见图3。[JP]
3含量测定
[JP3]31色谱条件色谱柱:Waters Xselect C18 (250mm × 46mm,5μm);[JP]流动相:甲醇-02%磷酸溶液(3∶97);检测波长270nm;柱温25℃;流速为10mL/min,进样量10μL。用紫外检测器检测。在此色谱条件下,没食子酸和其它组分均能達到分离。样品中没食子酸保留时间与对照品保留时间一致。见图4。
32供试品溶液的制备取供试品,剪碎,取约10g,精密称定,置具塞锥形瓶中,加50%甲醇溶液25mL,精密称定重量,超声处理30min,冷却至室温,再称定重量,用50%甲醇补足减失的重量,滤过,取续滤液,即得。
33对照品溶液的制备取没食子酸对照品257451mg,用50%甲醇溶解,定容于50mL的容量瓶中,得对照溶液05149mg /mL。
34线性范围的考察精密吸取没食子酸对照品溶液(05149mg/mL)各1、2、4、8、16、20μL,注入液相色谱仪,记录色谱图,得到相应的峰面积。以没食子酸对照品浓度为X坐标,以相应峰面积为Y坐标进行回归处理,得回归方程Y=3E+06X+44703,没食子酸线性范围为05149~10298μg,R2=09999 (n=6)。见图5。
35精密度试验精密吸取对照品溶液10μL,连续进样6次,计算精密度,结果6次测定值的RSD低于2%,表明该方法精密度良好。
36重复性试验精密吸取供试品溶液10μL,注入液相色谱仪,共6次,记录色谱图,6次测定值的相RSD低于2%,表明该方法重复性良好。
37稳定性试验精密吸取供试品溶液10μL,在上述色谱条件下在0、1、2、8、16、24h测得没食子酸的峰面积,计算其RSD低于2%,表明样品在24h内具有较好的稳定性。
38加样回收试验取已知含量的供试品(没食子酸的含量为53740mg/g)6份,分别精密加入对照品溶液(含没食子酸的量05149mg/mL)50mL,再照供试品溶液的制备法提取,按确定的测定方法进行测定,计算回收率,结果表明:回收率在95%~105%之间,该方法回收率良好。见表2。
39样品测定按上述提取方法和色谱条件,对7批次的甘露酥油丸进行了含量测定,测得每批样品中没食子酸的含量。以平均含量下浮20%作为本品的含量限度标准,故暂定本品每克没食子酸不得少于520mg/g。见表3。
4讨论
参考相关文献[4]诃子、毛诃子、余甘子的主要成分均为没食子酸,故采取高效液相色谱法测定没食子酸总量。
诃子、毛诃子、余甘子的薄层鉴别[2,5]。经过多次试验,在薄层色谱行为的重现性良好,专属性强,Rf值适中,分离效果好,因此可以将此方法定为甘露酥油丸的定性鉴别。
取没食子酸对照品溶液适量,采集200~400nm波长间紫外吸收光谱,发现其在270nm 有最大吸收。且没食子酸对照品色谱峰与其它峰达到良好的基线分离,杂质干扰少,峰形好。
参考文献
[1]卫生部药典委员会.中华人民共和国卫生部药品标准(藏药第一册)[S]. 1995.
[2]国家药典委员会.中华人民共和国药典(第一部) [S].北京:中国医药科技出版社,2015.
[3]国家药典委员会.中华人民共和国药典(第四部) [S].北京:中国医药科技出版社,2015:103.
[4]中华本草编委会.中华本草·藏药卷[M].上海:上海科技出版社, 2002.
[5]卫生部药典委员会. 中华人民共和国药典中药薄层色谱彩色图集[M].广州:广东科技出版社,1993.
(編辑:穆丽华)
