N-丁基-4-[乙基-(3-甲基苯基)氨基]丁酰胺的合成
2017-02-07丁成荣魏一凡刘宝国季国炎
丁成荣, 魏一凡, 刘宝国, 季国炎
(1. 浙江工业大学 化学工程学院,浙江 杭州 310014; 2. 绍兴东湖高科股份有限公司,浙江 绍兴 312074)
·研究简报·
N-丁基-4-[乙基-(3-甲基苯基)氨基]丁酰胺的合成
丁成荣1, 魏一凡1, 刘宝国1, 季国炎2*
(1. 浙江工业大学 化学工程学院,浙江 杭州 310014; 2. 绍兴东湖高科股份有限公司,浙江 绍兴 312074)
以γ-丁内酯为起始原料,经开环氯化和酰化反应制得N-丁基-4-氯丁酰胺(2); 2与N-乙基间甲苯胺经N-烷基化反应合成了N-丁基-4-[乙基-(3-甲基苯基)氨基]丁酰胺,总收率74.6%,纯度97.6%,其结构经1H NMR和MS(ESI)确证。
γ-丁内酯;N-丁基-4-氯丁酰胺;N-丁基-4-乙基-(3-甲基苯基)氨基]丁酰胺; 合成
N-丁基-4-[乙基-(3-甲基苯基)氨基]丁酰胺(3)是一种重要的精细化学品中间体,在分子生物学、微生物医学、感光材料与染料等领域具有广泛的用途[1-4]。目前,其作为杂环偶氮分散染料的重要中间体,可用于制备代替分散蓝56的低温型蓝色染料,获得色泽鲜艳、染色均匀、吸光度高、日晒牢度好的环保型杂环类分散染料[5-6],极大地解决了分散蓝56价格高和三废处理困难等问题[7-8],从而具有较大的经济价值和社会效益。
到目前为止,3的合成研究尚无报道,其类似结构的化合物N-(2-溴苯基)-4-[乙基-(3-甲基苯基)氨基]丁酰胺是以N-乙基间甲苯胺为起始原料,与4-溴丁酸乙酯发生N-烷基化反应得4-(乙基-3-甲基苯胺基)丁酸乙酯(17 h);后经水解反应(12 h)所得酸再与2-溴苯胺在缩合试剂1-羟基苯并三唑与1-乙基-3-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐作用下(17 h)制得最终产物[9]。该路线原料难以获得、反应时间长、操作流程复杂、成本较高、不利于工业化生产。
为了降低成本,满足工业化生产的需求,本文在参考文献[10]方法的基础上设计了以γ-丁内酯、正丁胺和N-乙基间甲苯胺为主要原料合成3的新工艺。以γ-丁内酯为起始原料,经开环氯化和酰化反应制得N-丁基-4-氯丁酰胺(2); 2与N-乙基间甲苯胺经N-烷基化反应合成了3,总收率74.6%,纯度97.6%,其结构经1H NMR和MS(ESI)确证。并对反应条件进行了探索与优化。
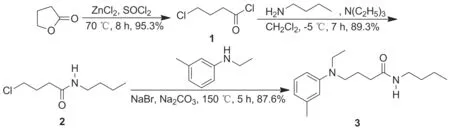
Scheme 1
1 实验部分
1.1 仪器与试剂
Bruker AVANCE III 500 MHz型核磁共振仪(CDCl3为溶剂,TMS为内标);Thermo Fisher ITQ 110型直接进样杆离子阱质谱仪;GC-2014型气相色谱仪。
γ-丁内酯,南通益同化工有限公司;N-乙基间甲苯胺,江苏仲鼎化工有限公司;其余所用试剂均为分析纯或化学纯。
1.2 合成
(1) 4-氯丁酰氯(1)的合成[11-13]
在三口瓶中加入γ-丁内酯43.0 g(0.50 mol)和无水氯化锌3.0 g(0.02 mol),搅拌下于60 ℃反应1 h;待反应液澄清透明后,缓慢滴加氯化亚砜65.4 g(0.55 mol),滴毕,于70 ℃反应8 h(反应产生的二氧化硫用30%NaOH溶液吸收)。减压蒸馏得刺激性无色液体1 67.8 g,收率95.3%(以γ-丁内酯计),含量99.1%(GC,下同);1H NMRδ: 3.62(t,J=6.2 Hz, 2H), 3.13(t,J=7.0 Hz, 2H), 2.24~2.11(m, 2H)。
(2) 2的合成
在三口瓶中,依次加入正丁胺24.1 g(0.33 mol),三乙胺30.3 g(0.3 mol)和二氯甲烷120 mL,控制温度为-5 ℃,缓慢滴加1 42.3 g(0.3 mol),滴毕(6 h),保温反应1 h。加水50 g,高速搅拌30 min,分液,有机相依次用稀盐酸(2×20 mL)和饱和食盐水(2×20 mL)洗涤,无水硫酸镁干燥,常压蒸除二氯甲烷(套用于下批)得淡黄色液体2 48.4 g,含量98.3%,收率89.3%(以1计);1H NMRδ: 6.75(s, 1H), 3.45(t,J=6.4 Hz, 2H), 3.08(dd,J=13.0 Hz, 7.1 Hz, 2H), 2.24(t,J=7.3 Hz, 2H), 2.02~1.86(t,J=7.5 Hz, 2H), 1.40~1.28(m, 2H), 1.20(m, 2H), 0.79(q,J=7.25 Hz, 3H); MS(ESI)m/z: 178.1{[M+H]+}。
(3) 3的合成
在三口瓶中加入2 53.3 g(0.30 mol),碳酸钠15.9 g(0.15 mol),N-乙基间甲苯胺44.6 g(0.33 mol)和溴化钠3.2 g,搅拌下于150 ℃反应5 h。减压蒸除过量N-乙基间甲苯胺得黏稠状3粗品,加入适量乙酸乙酯,剧烈搅拌10 min,抽滤除去无机盐,滤液减压蒸除乙酸乙酯得棕黄色液体3 74.5 g,含量97.6%,收率87.6%(以2计);1H NMRδ: 7.16~7.05(m, 1H), 6.52(m, 3H), 5.49(s, 1H), 3.38~3.24(m, 6H), 2.32(s, 3H), 2.23(t,J=7.0 Hz, 2H), 1.93(dd,J=14.5 Hz, 7.3 Hz, 2H), 1.50~1.44(m, 2H), 1.35(m, 2H), 1.15(t,J=7.0 Hz, 3H), 0.93(t,J=7.3 Hz, 3H); MS(ESI)m/z: 277.2{[M+H]+}。
2 结果与讨论
2.1 合成2的反应条件优化
为了寻找2的最佳合成条件,分别考察了溶剂、原料配比[r=n(正丁胺) ∶n(1)]、反应温度和反应时间对收率的影响。
(1) 溶剂
正丁胺0.33 mol,其余反应条件同1.2(2),考察溶剂对收率的影响,结果见表1。文献方法采用四氢呋喃作为酰化反应的溶剂,由于四氢呋喃在水中有一定的溶解度,所以为纯化产物需先减压脱溶除去四氢呋喃,残渣再溶于乙酸乙酯进行后处理。多溶剂的交叉大量使用必然带来回收套用和能耗问题,由于中间体2不溶于水,为简化后处理环节,本文选用与水不互溶的溶剂,反应完全后加水打浆溶解三乙胺盐酸盐,除水后即可得到中间体2。由表1可见,二氯甲烷作溶剂时,收率最高(89.3%),故以二氯甲烷为溶剂。

表1 溶剂对收率的影响*
*正丁胺 0.33 mol,其余反应条件同1.2(2)。
(2)r
二氯甲烷为溶剂,其余反应条件同2.1(1),考察r对收率的影响,结果见表2。从表2可知,随着正丁胺投料量增加,收率提高,当r为1.10时,收率达89.3%,继续增大r,收率没有明显提升,故该反应的最佳r为1.10。

表2 r对收率的影响*
*溶剂为二氯甲烷,其余反应条件同2.1(1)。
(3) 反应温度
r为1.10,其余反应条件同2.1(2),考察反应温度对收率的影响,结果见表3。该反应是放热反应,在滴加过程中会放出大量热,二氯甲烷沸点较低,因此低温使反应安全且易于控制。由表3可知,温度超过-5 ℃,反应会有大量杂质出现,经分析是因酰胺上的N仍具有一定亲核性,会与氯相连的碳原子在温度较高时发生亲核取代反应生成五元环状副产物,故控制合适的温度有助于降低杂质的生成,当反应温度为-5 ℃时,收率89.3%,反应温度低于-5 ℃时,需要较长的保温时间才能获得较高的收率,反应所需的能耗增大,故该反应的最佳温度为-5 ℃。

表3 反应温度对收率的影响*
*r=1.10,其余反应条件同2.1(2);a反应时间分别为7, 7, 7, 12, 12 h。
(4) 反应时间
反应温度为-5 ℃,其余反应条件同2.1(3),考察反应时间对收率的影响,结果见表4。结合实验现象及中控结果可知,反应时间是影响该酰化反应的重要因素。滴加速度过快时,会导致体系局部温度快速升高,从而使产物自身环合生成五元环杂质,且杂质难以有效除去。滴加速度过慢时,效率过低。当反应时间为7 h时,收率89.3%,继续增加滴加时间,收率变化不明显,且延长时间意味着能耗增大和生产周期延长,综合考虑,故采用滴加时间为6 h,保温反应1 h,总计反应7 h。

表4 反应时间对收率的影响*
*反应温度为-5 ℃,其余条件同2.1(3);b滴加时间分别为4, 5, 6, 7, 8 h。
综上所述,合成2的最佳反应条件为:正丁胺0.33 mol,二氯甲烷为溶剂,n(正丁胺) ∶n(1)=1.10,于-5 ℃反应7 h,收率89.3%。
2.2 合成3的反应条件优化
该反应属于芳胺N-烷基化反应[14],亲核取代活性与卤原子的种类有关,溴与碘相对于氯是更好的离去基团,能有效加快反应速率,故在氯化物的亲核取代反应中加入溴盐或碘盐作为催化剂能促进反应的进行,但KI价格昂贵,考虑到工业成本问题,故选用活性较高且经济易得的NaBr作为催化剂。分别考察了原料配比[γ=n(N-乙基间甲苯胺) ∶n(2)]、碱、催化剂用量、反应温度和反应时间对收率的影响。
(1)γ
2 0.3 mol,其余反应条件同1.2(3),考察γ对收率的影响,结果见表5。由于该反应采用无溶剂法,反应后期较为粘稠,当γ较低时,反应体系中由于传质等原因会造成中间体2与N-乙基间甲苯胺不能充分接触而反应不完全,收率较低,同时,未反应完的原料不易除去,亦影响产品纯度,故采用廉价易得的N-乙基间甲苯胺过量,促使中间体2反应完全,由表5知,γ=1.10时,反应收率已经较高(87.6%),考虑到工业化生产对原料成本的要求,最佳γ为1.10。

表5 γ对收率的影响*
*2 0.3 mol,其余反应条件同1.2(3)。
(2) 碱
该反应为亲核取代反应,每分子原料相互反应会生成一分子HCl,为加快反应速度,需要使用碱中和生成的HCl。γ为1.10,其余反应条件同2.2(1),考察不同碱对收率的影响,结果见表6。从表6可知,三乙胺和吡啶作为有机碱,长时间高温均相反应,生成了部分杂质,反应收率较低;NaOH和KOH碱性过强,长时间高温下反应增加了中间体2发生自身环合的几率;故采用碱性适中的无机碱Na2CO3作为缚酸剂。
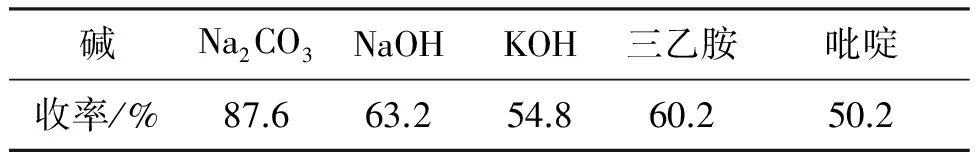
表6 碱对收率的影响*
*γ为1.10,其余反应条件同2.2(1)。
(3) 催化剂用量
碳酸钠为缚酸剂,其余反应条件同2.2(2),考察不用催化剂用量对反应的影响,结果见表7。由表7可知,随着催化剂用量的增大,溴离子的浓度升高,增加了双分子亲核取代反应的有效碰撞几率,收率逐渐提高。催化剂用量为6%时,收率达87.6%,继续增加催化剂用量,反应收率基本保持不变,故选用6%NaBr作为催化剂。
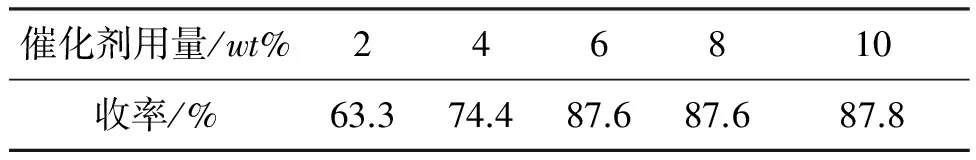
表7 催化剂用量对收率的影响*
*碳酸钠为缚酸剂,其余反应条件同2.2(2)。
(4) 反应温度
反应温度是主要的动力学参数,对反应速率有较大影响。6%NaBr作为催化剂,其余反应条件同2.2(3),考察不同反应温度对收率的影响,结果见表8。由表8可知,当温度低于150 ℃时,反应进行较慢,单位时间内反应收率较低,当温度为150 ℃时,收率最高(87.6%),当温度高于150 ℃时,过高的温度会引起副反应的发生,严重影响了产物浓度,故150 ℃为最佳反应温度。

表8 反应温度对收率的影响*
*6% NaBr为催化剂,其余反应条件同2.2(3);a反应时间分别为15, 15, 15, 8, 5, 5, 5 h。
(5) 反应时间
利用气相色谱分析跟踪测定中间体2的转化率确定反应终点,反应温度为150 ℃,其余反应条件同2.2(4),考察不同反应时间对收率的影响,结果见表9。由表9可知,随着反应时间的延长,收率有明显提高,当反应时间为5 h时,收率最高(87.6%)。继续延长反应时间,长时间高温下,产物会与反应生成的水发生水解反应,降低反应收率。故最佳反应时间为5 h。

表9 反应时间对收率的影响*
*反应温度为150 ℃,其余反应条件同2.2(4)。
综上所述,合成3的最佳反应条件为:碳酸钠为碱,NaBr 6wt%为催化剂,n(N-乙基间甲苯胺) ∶n(2)为1.10,于150 ℃反应5 h,收率87.6%。
3 结论
以γ-丁内酯为起始原料,经3步反应合成了N-丁基-4-[乙基-(3-甲基苯基)氨基]丁酰胺,总收率74.6%,纯度97.6%。该合成路线原料价廉易得,操作简单,经济环保,满足工业化生产要求,具有一定的应用前景。
[1] Ewing G, Mullah K, Graham R. Non-fluorescent quencher compounds and biomolecular assays:WO 03019145[P].2003.
[2] Cellier M, James A L, Orenga S,etal. Novel chromogenic aminopeptidase substrates for the detection and identification of clinically important microorganisms[J].Bioorganic & Medicinal Chemistry,2014,22(19):5249-5269.
[3] Taniguchi M, Ooki N, Nakamura K. Developing agent for photograhy,processing composition containing this agent and color image forming method using this composition:JP H03246543 A[P].1991.
[4] 陆建焕,谢毅,方东. 一种偶氮型杂环分散染料:CN 102898856 A[P].2013.
[5] 陈荣圻. 分散染料六十年发展概述(二)[J].染料与染色,2015,43(1):18-30.
[6] 梁秋雯,徐戈凯,邹盼盼,等. 近10年有关分散染料研究的一些进展[J].染料与染色,2014,(5):13-20.
[7] 赵国生,陈宝兴,陈田木,等. 分散蓝56缩合母液处理及资源回收[J].化工管理,2015,34:161-162.
[8] 耿宁宁,杨军海,荆丽丽. 两支替代C.I.分散蓝56的分散染料应用探讨[J].针织工业,2016,(1):47-50.
[9] Nakagawa T, Suzuki T. Amide derivative:US 8 168 827[P].2012.
[10] Takao K, Noda K, Morita Y,etal. Molecular and crystal structures of uranyl nitrate complexes withN-alkylated 2-pyrrolidone derivatives:Design and optimization of promising precipitant for uranyl ion[J].Crystal Growth & Design,2008,8(7):2364-2376.
[11] 丁成荣,李伟华,魏一凡,等. 4-(N-乙基-3-甲基苯胺基)丁酸甲酯的合成[J].合成化学,2016,24(6):529-532.
[12] 牛宇岚,李敏,段海龙. 4-氯丁酰氯的合成研究[J].精细化工中间体,2006,36(3):21-22.
[13] Juraj Galeta, Lukás Tenora. Dihydropyrrolo[1,2-b]pyrazoles:Withasomnine and related compounds[J].Tetrahedron,2013,69(34):7139-7146.
[14] 师华,陆峰,熊家锦,等.N-烷基化反应[J].精细化工中间体,2008,38(6):8-11.
[15] 凌青,李欣,沈竞康. 仲胺的合成方法新进展[J].合成化学,2007,15(3):247-253.
Synthesis ofN-Butyl-4-[ethyl(3-methylphenyl)amino] Butanamide
DING Cheng-rong1, WEI Yi-fan1, LIU Bao-guo1, JI Guo-yan2*
(1. College of Chemical Engineering, Zhejiang University of Technology, Hangzhou 310014, China; 2. Shaoxing Donghu High-tech Co., Ltd., Shaoxing 312074, China)
N-butyl-4-[ethyl(3-methylphenyl)amino]-butanamide with the total yield of 74.6% and the purity of 97.6% was synthesized by the reaction ofN-ethyl-3-toluidine withN-butyl-4-chlorobutanamide, which was synthesized through ring-opening chlorination and acylation reaction using 1,4-butyrolactone as the starting material. The structure was confirmed by1H NMR and MS(ESI).
1,4-butyrolactone;N-butyl-4-chlorobutanamide;N-butyl-4-[ethyl(3-methylphenyl)amino] butanamide; synthesis
2016-07-28; 修改日期: 2016-10-25
丁成荣(1966-),男,汉族,浙江东阳人,教授,主要从事有机合成的研究。 E-mail: dingcr@zjut.edu.cn
季国炎,工程师, Tel. 0575-88038001, E-mail: 103783895@qq.com
O625.63
A
10.15952/j.cnki.cjsc.1005-1511.2017.01.16197
