Bi/AlSb(110)体系的结构和电子性质的第一性原理研究
2017-01-20文黎巍裴慧霞
文黎巍,裴慧霞
(1.河南工程学院 理学院,河南 郑州 451191; 2.郑州铁路职业技术学院 电气工程系,河南 郑州 451460)
Bi/AlSb(110)体系的结构和电子性质的第一性原理研究
文黎巍1,裴慧霞2
(1.河南工程学院 理学院,河南 郑州 451191; 2.郑州铁路职业技术学院 电气工程系,河南 郑州 451460)
使用第一性原理密度泛函理论研究了在AlSb(110)衬底表面外延生长Bi薄膜的表面结构和电子性质,计算了生长1~6层Bi薄膜的层间距、吸附能与能带结构.研究发现,不同层数的Bi薄膜显示出奇偶振荡:奇数层稳定,偶数层不稳定; 1,3奇数层为半导体,2,4偶数层为金属,5,6层均为金属态,不存在振荡.
第一性原理;Bi薄膜;吸附能
近年来,V族元素在半导体衬底上,特别是在GaAs(110)衬底表面的外延生长,在理论和实践中都得到了广泛的研究[1-6].这些研究主要关注外延生长薄膜的结构、低能电子衍射的动力学分析、薄膜的诱导重组等问题.Bi掺杂的GaAs还可以用来做饱和吸收器,以提高半导体激光器的发光功率和单脉冲能量等.Bi薄膜在III-V族元素AsGa,InP,InAs等衬底的(110)表面上生长研究已有报道[7-10],理论计算得到单层有序生长的几何和电子能带结构,与实验上扫描隧道显微镜、角分辨光电子能谱等研究结果吻合较好.但是,AlSb基Bi薄膜结构和稳定性仍然未知.本研究用基于密度泛函理论(DFT)的Vienna Ab-initio Simulation Package(VASP)程序包[11-12]计算,对AlSb(110)衬底外延生长Bi薄膜的结构和稳定性进行了研究,分别计算了外延生长1~6层Bi薄膜的层间距离、吸附能、能带结构和带隙.结果表明,不同层数的稳定性和材料导电性存在奇偶振荡的转变,分析了其形成原因并找出了规律.
1 计算方法
本研究采用基于密度泛函理论的平面波展开方法,原子核区域用超软赝势描述,交换关联能选用的是局域密度近似(LDA).模型构建基于周期性边界条件的平板模型,在8层AlSb衬底两侧分别生长1~6层的Bi薄膜.Bi薄膜的初始位置依据外延生长结构模型给定,结构弛豫过程中力的收敛标准为0.02 eV/Å,布里渊区K点数为9×9×1形式.
2 结果与讨论
2.1 单层Bi薄膜
图1给出了纯净的AlSb(110)衬底表面和在AlSb(110)衬底表面外延生长一层Bi薄膜的结构,其中(a)和(b)分别表示纯净的AlSb(110)衬底表面弛豫前和弛豫后的结构.对比图1(a)和图1(b)可以看出,弛豫后最顶层的Al原子向里移动,倾向于sp2杂化,Sb原子向外移动,倾向于sp3杂化,这与化学键旋转模型相对应.图1(c)给出了在AlSb(110)衬底表面外延生长一层Bi薄膜的侧视图,可以看出最顶层的Al原子仍旧停留在块体时的位置.图1(d)给出了外延生长的俯视图,点线指出了表面原胞.图2给出了Bi/AlSb(110)系统的电荷密度图,图中(a)和(c)分别对应Bi原子和衬底表面的Al和Sb原子之间的电荷密度,(b)和(d)是与之对应的差分电荷密度.从电荷密度图中可以看出,Bi原子与Al和Sb原子有较强的化学结合,差分电荷密度图表面的Bi原子与Al原子之间有明显的电荷转移,成共价键,而Bi原子与Sb原子之间的电荷转移不明显.这是由于Bi是Sb的同族原子,在AlSb表面生长后与Al成化学键结合,与Sb成金属键,表面结构呈现清晰的外延生长结构.
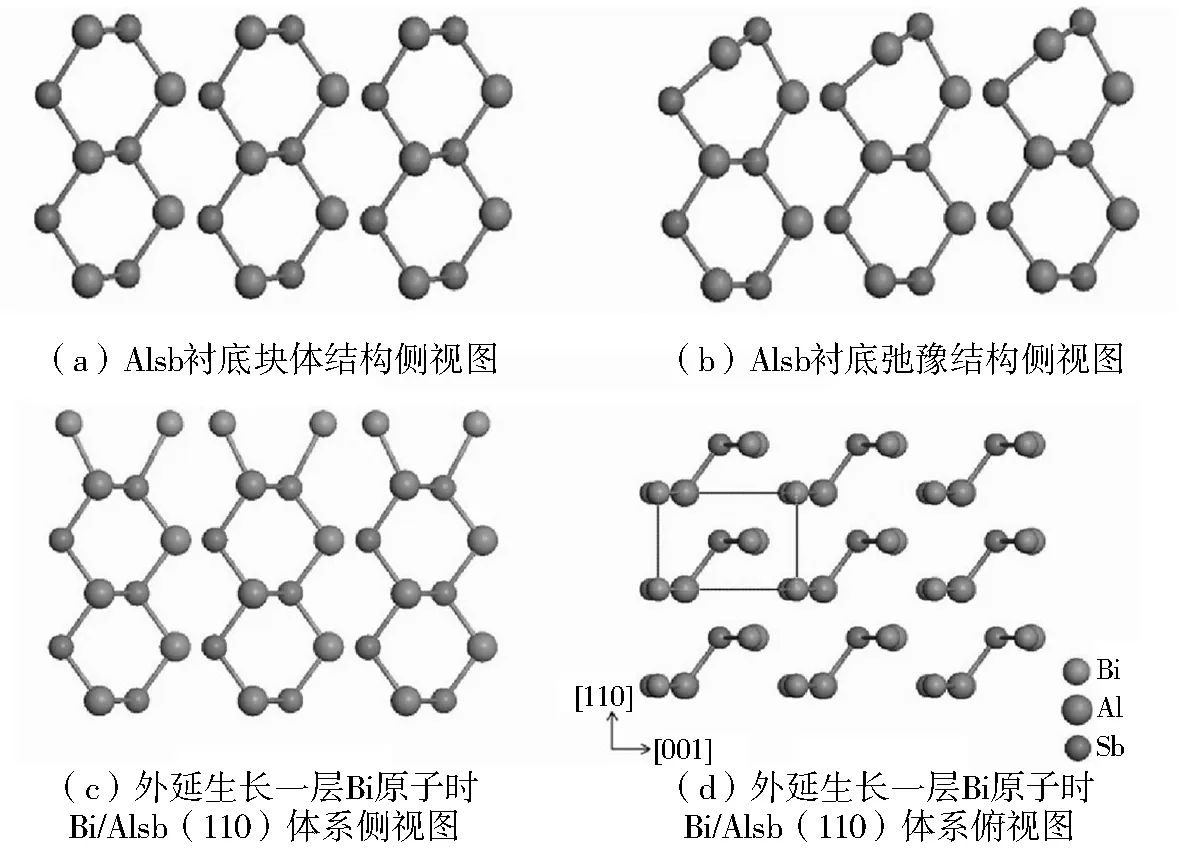
图1 AlSb(110)衬底表面外延生长1层Bi薄膜的结构图Fig.1 Structure of one layer Bi/AlSb(110) substrate

图2 Bi/AlSb(110)系统的电荷密度图Fig.2 Charge density contours for ECL structures of Bi/AlSb(110) system
2.2 多层Bi薄膜
为了研究Bi/AlSb(110)系统随着Bi层数增加的相对稳定性,通过公式-Ead=[E(n)-E(n-1)-4EBi]÷4计算了每个Bi原子的能量(Ead),其中E(n)和 E(n-1)分别表示第n层和n-1层的总能量,EBi表示孤立的Bi原子的总能量.当n=1时,E(n-1)表示纯净的AlSb(110)表面的总能量.

图3 Bi/AlSb(110)体系不同层数最高界层距离和吸附能Fig.3 The oscillation in the adhesion energy to add a new layer of Bi to AlSb is compared to similar oscillations seen in the topmost interlayer distance
不同层数吸附能的负值和对应层数薄膜的最外层层间距离见图3,吸附能显示出强烈的尺寸效应振荡,在1,3,5,7层的吸附能比2,4,6层大.从这些数据可以看出,衬底上有单层Bi薄膜时的吸附能最大,这时键与衬底结合最强烈.外延生长薄膜在3,5,7层时局部稳定,而在2,4,6层时不稳定,这决定了薄膜的双层生长模式.从图3还可以看出,吸附能在5层以上时随着层数的增加变化不大,这说明在5层以上生长模式可能会发生变化.
这种与薄膜层数有关的震荡变化被认为是量子尺寸效应的结果.例如,以往的Sb 在GaAs(110)衬底外延生长的第一性原理研究中,吸附能的起伏振荡就归结于Bi薄膜的量子尺寸效应.在图3中,同时给出了吸附能和层间距离,可以看到它们相符得很好.对于奇数层薄膜,最外层层间距小、吸附能较大;对于偶数层薄膜,最外层层间距大、吸附能较小.图3给出的最高界层距离和计算出来的吸附能相符很好,这种奇偶震荡不止源于量子尺寸效应,还与薄膜最外层层间距有关.第1层Bi薄膜与衬底化学吸附结合,与衬底之间只有2.5 Å;从第2层薄膜开始,Bi薄膜层间距震荡变化,1层和2层薄膜层间距大,2层和3层薄膜层间距小,显示了明显的双层生长模式.吸附能的震荡也可以用层间距的变化解释,除去第1层与衬底化学结合Bi原子薄膜,当Bi薄膜层数(n-1)为奇数时,最外层Bi原子有未饱和悬挂键,吸附能小,而薄膜层数(n-1)为偶数时,Bi薄膜双层结合饱和悬挂键,吸附能大,结构稳定.
为了进一步了解薄膜结构稳定性的奇偶振荡转变对电子性质的影响,分别计算了AlSb(110)衬底表面外延生长1~6层Bi薄膜的能带结构,如图4所示.从图4中可以很明显地看到,除了外延生长1层和3层Bi薄膜呈现非金属性质外,其他生长层均显现出金属性.下面对每一层的能带进一步分析,首先看生长1层Bi薄膜的非金属性能带,主体带隙间有4条子能带,两条满带能量低,两条空带能量高,计算出带隙为0.5 eV.图4(b)外延生长2层Bi薄膜金属能带图中,主体带隙间增加了2条子能带且交叠横穿费米能级.这些横穿费米能级的子能带随着Bi薄膜厚度的增加出现振荡,在图4(c)中消失而又在图4(d)中出现.1层和3层Bi薄膜显示出非金属性,其带隙分别为0.5 eV和0.14 eV,4层和5层及以上都显示出金属性,此结果与薄膜结构的稳定性相符.

图4 AlSb(110)衬底表面外延生长1~6层Bi薄膜的能带图Fig.4 Surface band structures of Bi/AlSb(110)-(1×1) at six Bi coverages
2.3 讨论
以上研究表明,AlSb(110)基外延生长的Bi薄膜显示出结构稳定性和电子性质由金属性和半导体性质的奇偶振荡转变,在4层以上实现了半导体-金属的过渡,本结论与实验上发现的非金属-金属的转变和Bi薄膜的厚度有关的结论一致.这种转变可以用量子力学中的量子阱模型来解释:由于与衬底接触的第1层Bi原子与衬底作用较强,认为量子阱从第2层开始,每个Bi原子的配位数是4,然而一个Bi原子具有6个价电子,故外延生长的每个Bi原子具有2个自由电子.对于1×1的单元晶胞,每层有1个Bi原子对应2个自由电子,所以总共的自由电子数目可设为2n,n=1,2,3,…,对应层数为2,3,4.从另一方面说,每增加一层,就出现2个子能带,不同层数的Bi原子对应形成不同的子能带.当薄膜厚度非常薄且只有几个原子层时,不同层的原子形成的子能带不存在相互交叠,可以出现金属-非金属的转变——偶数层是金属,奇数层是非金属.因此,第2,4层为金属,第3层为非金属.当薄膜超过临界厚度时,不同层数的原子形成的子能带相互交叠,振荡消失,实现了金属性的转变.这种震荡也与Bi薄膜的双层生长模式有关,第一层Bi薄膜与衬底化学结合,没有悬挂键,结构稳定,具有半导体性质;再生长一层Bi薄膜,没有形成双层结构,有悬挂键,结构不稳定,显示金属性质;第3层Bi薄膜结构,后面两层也形成双层结构,没有悬挂键,结构稳定,显示半导体性质.
3 结论
用第一性原理密度泛函理论研究了在AlSb(110)衬底上外延生长不同层Bi薄膜体系的结构和能带.结果发现,外延生长奇数层时稳定,偶数层时不稳定,引起吸附能与非金属-金属性质随薄膜层数的改变而产生奇偶振荡变化;在4层以后,实现了非金属到金属的转变,这是由薄膜的量子尺寸效应与薄膜的双层生长模式共同引起的.
[1] CONG W,LI D C,ZHAO S Z,et al.Diode-pumped passively Q-switched Nd:GGG laser with a Bi-doped GaAs semiconductor saturable absorber[J].Optics Communications,2014(332):292-295.
[2] PUNKKINEN M P J,LAUKKANEN P,KUZMIN M,et al.Does Bi form clusters in GaAs1-xBixalloys [J].Semiconductor Science and Technology,2014,29(11):115007.
[3] GUO T,ATKINSON R E,FORD W K.Growth of bismuth films on GaAs (110) studied using low-energy electron diffraction[J].Physical Review B,1990,41(8):5138.
[4] MIWA R H,TAKAHASHI E K.Bi-covered InAs (110)surfaces:an ab initio study[J].Surface Science,2004(566/568):949-955.
[5] GAY S C A,SRIVASTAVA G P.Ab initio investigation of Bi-covered GaSb(110) surfaces[J].Physical Review B,2000(61):2688.
[6] GEMMEREN T V ,LOTTERMOSER L,FALKENBERG G,et al.Bismuth-induced restructuring of the GaSb(110)surface[J].Physical Review B,1998,57(7):3749.
[7] SCHMIDT W G,BECHSTEDT F,SRIVASTAVA G P.Adsorption of group-V elements on III-V(110) surfaces[J].Surface Science Reports,1996(25):141-223.
[8] UMERSKI A,SRIVASTAVA G P.Geometry and electronic band structure of an ordered monolayer deposition of Bi on III-V(110) semiconductor surfaces[J].Physical Review B,1995(51):2334.
[9] PATRIN J C,LI Y Z,CHANDER M,et al.Sb and Bi on GaAs(110):Substrate-stabilized overlayer structures studied with scanning tunneling microscopy[J].Physical Review B,1992(46):10221.
[10]GEMMEREN T,LOTTERMOSER L,FALKENBERG G,et al.Growth morphology and structure of bismuth thin films on GaSb(110) [J].Surface Science,1998,414(1/2):254-260.
[11]KRESSE G,FURTHMULLER J.Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set [J].Physical Review B,1996(54):11169.
[12]VANDERBILT D.Soft self-consistent pseudopotentials in a generalized eigenvalue formalism[J].Physical Review B,1990(41):7892.
First principles study on structure and stability of Bi/AlSb(110) systems
WEN Liwei1,PEI Huixia2
(1.CollegeofSciences,HenanUniversityofEngineering,Zhengzhou451191,China;2.ElectricalEngineering,ZhengzhouRailwayVocationalandTechnicalCollege,Zhengzhou451460,China)
Using first-principle’s density functional theory,we investigate the structure and charge density of monolayer Bi film on AlSb(110)surface. The interlayer distance ,adhesion energy,and band gap of 1~6 monolayers Bi films exist odd even oscillation. The odd monolayers are stable and even monolayers are instable,the 1 and 3 odd monolayers are semiconductors,while 2 and 4 even monolayers are metals,5 and more monolayers are always metal without oscillation.
first principles;Bi film;adsorption energy
2016-04-06
河南省科技攻关计划项目(132102210141)
文黎巍(1981-),女,河南邓州人,讲师,研究方向为计算物理.
O469
A
1674-330X(2016)04-0079-04
