周氏回生丸质量标准提高研究
2017-01-16周军,王杰
周 军,王 杰
(天津市药品检验所,天津 300070)
周氏回生丸质量标准提高研究
周 军,王 杰
(天津市药品检验所,天津 300070)
目的:提高周氏回生丸的质量标准。方法:采用显微鉴别法对周氏回生丸中的五倍子、雄黄、朱砂进行定性鉴别;采用薄层色谱法对周氏回生丸中的木香、人工麝香、甘草进行定性鉴别;采用高效液相色谱法测定五倍子中没食子酸的含量,色谱柱为安捷伦ZORBAX TC-C18(250 mm×4.6 mm,5 μm),流动相为甲醇-0.1%磷酸(10∶90),柱温40 ℃,流速为0.8 ml/min,检测波长为273 nm。结果:显微鉴别中五倍子、雄黄、朱砂的显微特征明显,薄层色谱中木香、人工麝香、甘草的特征斑点明显,重现性好,无干扰;没食子酸在49.6~1 984.0 μg范围内线性关系良好, 五倍子水解前平均回收率为102.0%,RSD为1.30%(n=6),五倍子水解后平均回收率为99.34%,RSD为1.56%(n=6)。结论:本法专属性强、准确、快速,使产品的质量得到有效的控制,保证了临床疗效。
周氏回生丸,五倍子,雄黄,朱砂,木香,人工麝香,甘草,显微鉴别,薄层色谱,没食子酸,高效液相色谱法
周氏回生丸处方来源于明代紫金锭方加减,最早收载在1978年版《天津市中成药规范》、1983年《北京市药品标准》、1990年版《天津市药品标准》及卫生部《药品标准中药成方制剂第十七册》中;本品为《中国药典》2010年版一部增订品种,同时亦为国家药品标准提高行动计划中成药品种增修订品种。根据国家药品标准提高行动计划中成药品种增修订项目任务表的要求,对周氏回生丸质量标准进行了提高、完善,增加处方中五倍子、雄黄和朱砂的显微鉴别方法,增加了木香、甘草及人工麝香的薄层鉴别方法,并制定了五倍子的含量测定方法。修订后的质量标准提高了药品的质量控制指标,具有专属性强、准确度高的特点,现该标准已收录在《中国药典》2010年版一部及《中国药典》2015年版一部。
1 实验材料
1.1 仪器 岛津LC-2010C高效液相色谱仪,配有四元泵、紫外检测器、自动进样器、柱温箱、CLASS-VP工作站。METTLER TOLEDO AG135型电子天平(瑞士梅特勒-托利多集团生产);CAMAG薄层色谱成像系统(瑞士卡玛公司生产);薄层色谱用硅胶G板(烟台市化学工业研究所生产)。
1.2 试药 木香对照药材(批号120921-200502)、甘草对照药材(批号120904-200511)、麝香酮对照品(批号110719-200512)、没食子酸对照品(批号110831-200302,供含量测定用),均于中国药品生物制品检定所购置。甲醇(色谱纯,天津市康科德科技有限公司),盐酸、磷酸均为分析纯,水为去离子水。周氏回生丸样品由天津中新药业集团股份有限公司乐仁堂制药厂(批号0805001,0805002,0805003)、北京同仁堂科技发展股份有限公司制药厂(批号8040542)提供。
2 方法与结果
2.1 显微鉴别 鉴于本品为生粉入药,采用显微鉴别法对处方中五倍子、雄黄和朱砂进行鉴别,参照《中国药典》有关内容,制定了周氏回生丸显微鉴别标准。具体内容为:非腺毛1至数个细胞,有的顶端稍弯曲(五倍子);不规则碎块金黄色或橙黄色,有光泽(雄黄);不规则细小颗粒暗棕红色,有光泽,边缘暗黑色(朱砂)。经检验,四批样品均具有周氏回生丸的显微特征,符合规定。结果见图1。
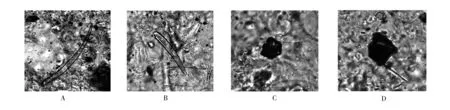
图1 五倍子的非腺毛(A、B) 雄黄的不规则碎块(C)朱砂的不规则细小颗粒(D)显微鉴别图谱
2.2 薄层色谱鉴别
2.2.1 木香的TLC鉴别 取本品适量,研细,取10 g,加50%甲醇60 ml,超声处理30 min,离心,取上清液,加石油醚(30~60 ℃)提取2次,每次20 ml,合并石油醚液,挥干溶剂,残渣加乙酸乙酯1 ml使溶解,作为供试品溶液[1,2]。另取木香对照药材0.25 g,加乙醚20 ml,超声处理10 min,滤过,滤液挥干,残渣加乙酸乙酯2 ml使溶解,作为对照药材溶液。按处方配比,取处方药材10 g(木香除外)按制法制得阴性样品,再按照供试品溶液制备方法处理,作为阴性样品溶液。按照《中国药典》2010年版一部TLC法试验,吸取供试品溶液4~10 μl、对照药材溶液2 μl、阴性对照溶液10 μl,分别点于同一硅胶G薄层板上,以正己烷-三氯甲烷-丙酮(6.5∶3.5∶0.1)为展开剂,展开,取出,晾干,喷以5%香草醛硫酸溶液,加热至斑点显色清晰。结果供试品色谱中在与对照药材色谱相应的位置上,显相同颜色的斑点,阴性样品色谱在相应位置无干扰,见图2。
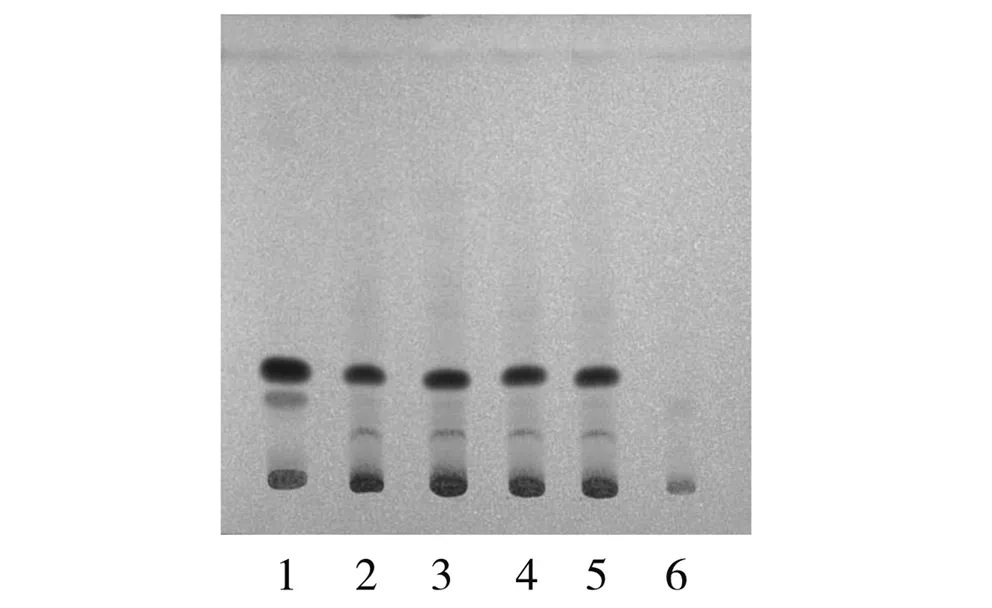
1.木香对照药材 2.样品1(批号0805001) 3.样品2(批号0805002) 4. 样品3(批号0805003) 5.样品4(批号8040542) 6.阴性样品
2.2.2 人工麝香的TLC鉴别
2.2.2.1 先加显色剂的方法 取“2.2.1”项下木香供试品溶液加入二硝基苯肼乙醇试液1 ml,置具塞试管中40 ℃温浸30 min,放冷,作为供试品溶液。另取麝香酮对照品,加乙酸乙酯制成每1 ml含2 mg的溶液,取1 ml,加二硝基苯肼乙醇试液1 ml,40 ℃温浸30 min,放冷,作为对照品溶液。再按处方配比,取处方药材10 g(人工麝香除外),按制法制得阴性样品,再按照供试品溶液制备方法处理,作为阴性样品溶液。按照《中国药典》2010年版一部TLC法试验,吸取供试品溶液及阴性对照溶液各10 μl、对照品溶液4 μl,分别点于同一硅胶G薄层板上,以石油醚(60~90 ℃)-二氯甲烷(2∶2)为展开剂,展开,取出,晾干。供试品色谱中,在与对照品色谱相应的位置上显相同颜色的斑点,阴性样品色谱在相应位置无干扰,见图3。
2.2.2.2 后喷显色剂的方法 取“2.2.1”项下木香供试品溶液作为供试品溶液。另取麝香酮对照品,加乙酸乙酯制成每1 ml含2 mg的溶液,作为对照品溶液。再按处方配比,取处方药材10 g(人工麝香除外),按制法制得阴性样品,再按照供试品溶液制备方法处理,作为阴性样品溶液。按照薄层色谱法《中国药典》2010年版一部TLC法试验,吸取供试品溶液及阴性对照溶液各10 μl、对照品溶液4 μl,分别点于同一硅胶G薄层板上,以石油醚(60~90 ℃)-二氯甲烷(2∶2)为展开剂,展开,取出,晾干,喷以二硝基苯肼乙醇试液。供试品色谱中,在与对照品色谱相应的位置上显相同颜色的斑点,阴性样品色谱在相应位置无干扰,见图3。
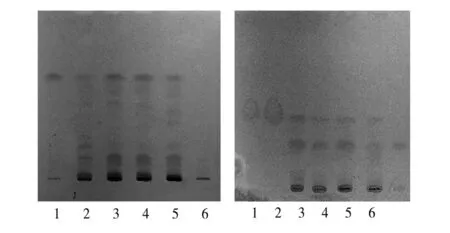
先加显色剂 后喷显色剂
2.2.3 甘草的TLC鉴别 取“2.2.1”木香鉴别项下石油醚提取后的50%甲醇溶液,蒸干,残渣加水15 ml使溶解,用乙酸乙酯提取2次,每次20 ml,合并乙酸乙酯液,蒸干,残渣加乙酸乙酯1 ml使溶解,作为供试品溶液[3]。另取甘草对照药材0.5 g,加50%甲醇溶液25 ml,超声处理30 min,滤过,滤液蒸干,残渣加水15 ml使溶解,用乙酸乙酯提取2次,每次20 ml,合并乙酸乙酯液,蒸干,残渣加乙酸乙酯1 ml使溶解,作为对照药材溶液。再按处方配比,取处方药材10 g(甘草除外),按制法制得阴性样品,再按照供试品溶液制备方法处理,作为阴性样品溶液。按照《中国药典》2010年版一部TLC法试验,吸取供试品溶液2~6 μl、对照药材溶液4 μl、阴性对照溶液6 μl,分别点于同一硅胶G薄层板上,以甲苯-乙酸乙酯-甲酸(14∶7∶2)为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,加热至斑点显色清晰,分别置日光及紫外光灯(365 nm)下检视。供试品色谱中,在与对照药材色谱相应的位置上,分别显相同颜色的斑点及荧光斑点,阴性对照溶液无干扰。结果见图4。

日光 UV(365 nm)
1.甘草对照药材 2.样品1(批号0805001) 3.样品2(批号0805002) 4.样品3(批号0805003) 5.样品4(批号8040542) 6.阴性样品溶液
图4 甘草薄层色谱图
3 没食子酸的测定
3.1 色谱条件 色谱柱为安捷伦TC-C18(250 mm×4.6 mm,5 μm);流动相:甲醇-0.1%磷酸(10∶90);柱温:40 ℃;流速:0.8 ml/min;检测波长:273 nm。理论板数按没食子酸峰计算不低于5 000。
3.2 溶液的制备及专属性试验
3.2.1 对照品溶液的制备 取没食子酸对照品适量,精密称定,加50%甲醇制成每1 ml含没食子酸30 μg的溶液,即得。
3.2.2 供试品溶液的制备 取同一批号(0805001)样品适量,研细,取0.5 g,精密称定,精密加入50%甲醇溶液50 ml,称定重量,置水浴中加热回流1.5 h,放冷,再称定重量,用50%甲醇补足减失的重量,离心,取上清液作为测定游离没食子酸的供试品溶液;再精密量取上清液5 ml,置圆底烧瓶中,减压蒸干,加入盐酸溶液(36→100) 15 ml,置沸水浴中加热2 h,放冷,转移至50 ml量瓶中,加50%甲醇至刻度,摇匀,作为测定总没食子酸供试品溶液。
3.2.3 阴性样品溶液的制备 按处方取除五倍子的各味药材,按制法制得阴性样品,再按“3.2.2”项下供试品溶液制备方法制得阴性样品溶液。
3.2.4 专属性试验 分别精密吸取对照品溶液10 μl、供试品溶液5 μl与阴性样品溶液5 μl,注入液相色谱仪,测定,结果阴性样品无干扰,见图5。
3.3 标准曲线的制备 取没食子酸对照品适量,精密称定,加50%甲醇分别制成每1 ml中含没食子酸4.96、9.92、19.84、39.68、99.20和198.40 μg的对照品溶液,分别精密吸取各10 μl,注入液相色谱仪,按“3.1”项下色谱条件分析,分别测定各自峰面积,以对照品进样量(μg)为横坐标,峰面积值为纵坐标,求得回归方程:Y=3 758X+12 438(r=0.999 9)。结果表明没食子酸在49.6~1 984.0 μg范围内线性良好。
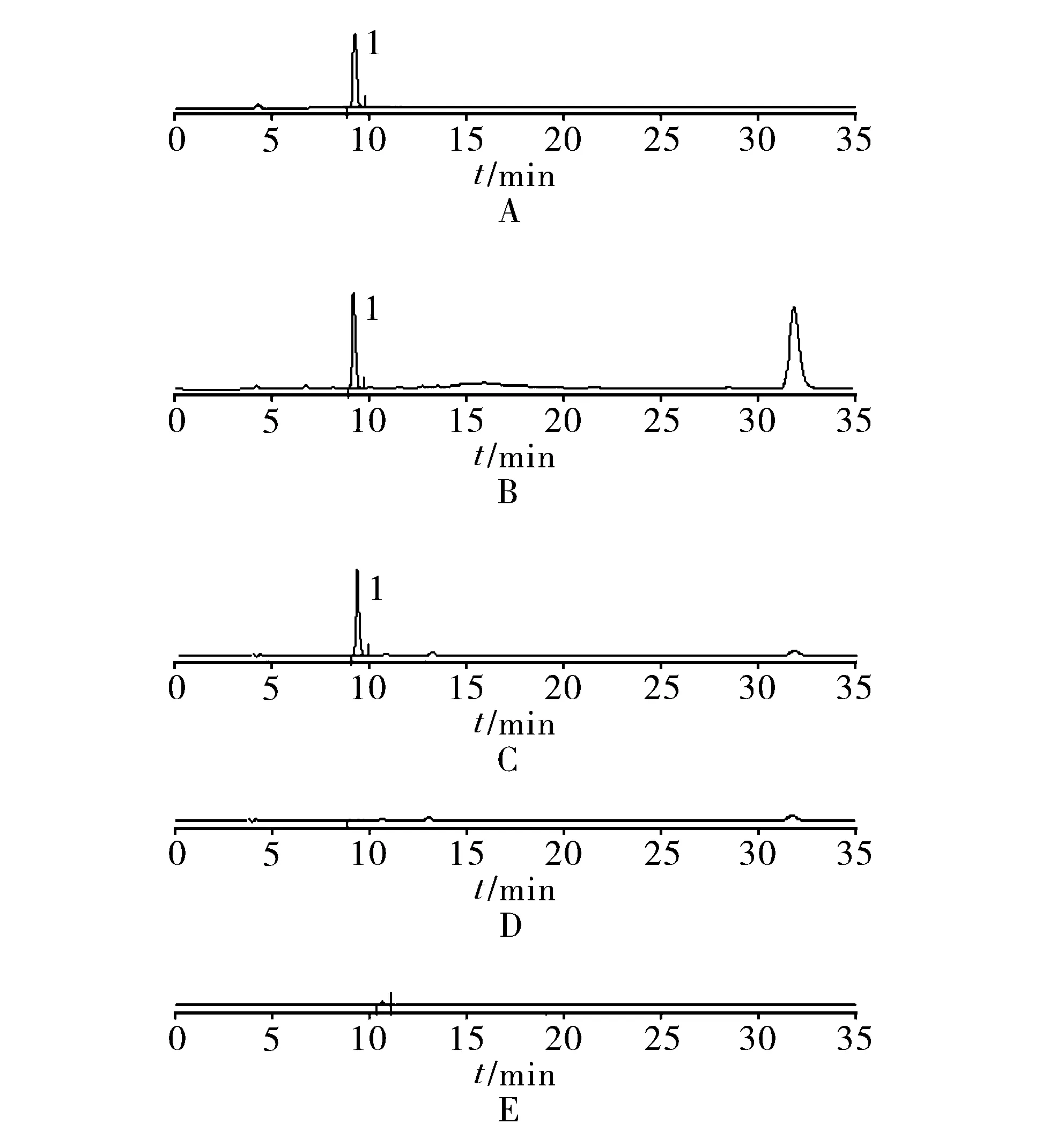
1.没食子酸
3.4 精密度试验 取同一批号样品(0805001)适量,研细,取约0.5 g,精密称定,按照“3.2.2”项下供试品溶液制备操作,按“3.1”项下色谱条件分析,连续进样6次,测定峰面积,水解前样品中没食子酸峰面积平均值为1 709 347.7,RSD为0.11%;水解后样品中没食子酸峰面积平均值为1 587 674.3,RSD为0.16%。
3.5 重复性试验 取同一批号样品(0805001)适量,研细,取约0.5 g,精密称定,共6份,按照“3.2.2”项下供试品溶液制备操作,按“3.1”项下色谱条件分析,测定,水解前样品中没食子酸含量平均值为8.887 1 mg/g,RSD为0.49%;水解后样品中没食子酸含量平均值为81.792 1 mg/g,RSD为0.93%。
3.6 稳定性试验 取同一批号样品(0805001)适量,取约0.5 g,精密称定,按照“3.2.2”项下供试品溶液制备操作,按“3.1”项下色谱条件分别在0、2、4、8、12、18和24 h进样,测定,水解前样品中没食子酸峰面积平均值为17 21 419.9,RSD为0.88%;水解后样品中没食子酸峰面积平均值为1 569 602.9,RSD为1.18%,表明两种供试品溶液在24 h内稳定。
3.7 加样回收率试验
3.7.1 水解前样品加样回收率 取同一批号样品(0805001)适量,取约0.25 g,精密称定,共6份,分别精密加入每1 ml中含55.58 μg的没食子酸对照品的50%甲醇溶液50 ml,按照“3.2.2”项下供试品溶液制备操作,至“离心,取上清液”,过微孔滤膜,制得供回收率用供试品溶液,按“3.1”项下色谱条件分析,计算回收率,结果水解前没食子酸平均回收率为102.0%,RSD为1.30%。结果见表1。
3.7.2 水解后样品加样回收率 取同一批号样品(0805001)适量,取约0.25 g,精密称定,共6份,分别精密加入每1 ml中含408.62 μg的没食子酸对照品的50%甲醇溶液50 ml,按照“3.2.2”项下供试品溶液制备操作,制得供回收率用供试品溶液,按“3.1”项下色谱条件分析。计算回收率,结果没食子酸平均回收率为99.34%,RSD为1.56%。结果见表2。
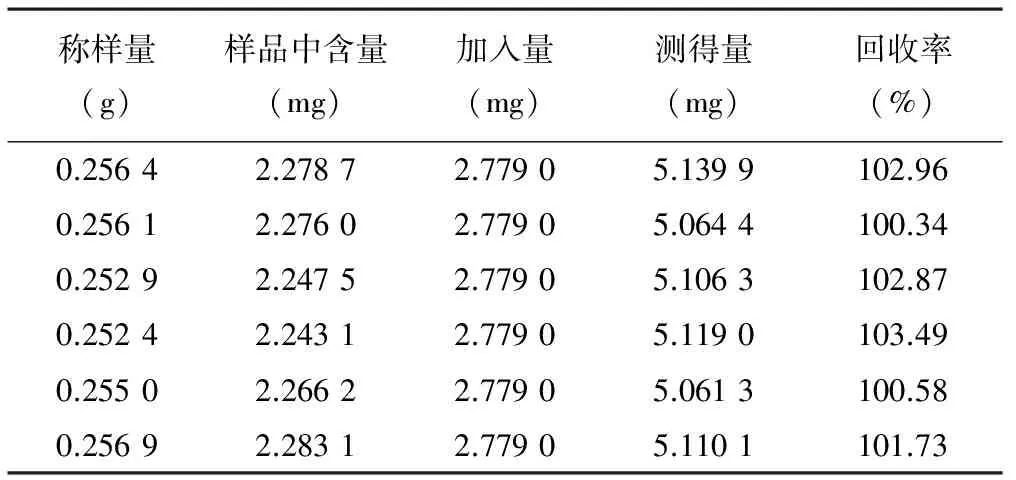
表1 水解前没食子酸加样回收率试验结果(n=6)
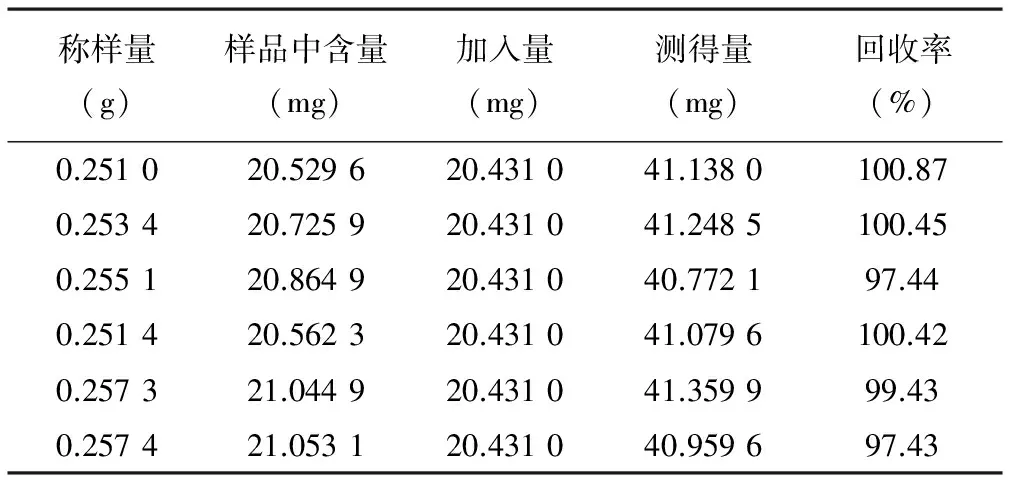
表2 水解后没食子酸加样回收率试验结果(n=6)
3.8 样品测定 取2个厂家4个批号的样品,按照“3.2.2”项下供试品溶液制备操作,按“3.1”项下色谱条件分析,测定,计算样品中没食子酸含量。结果见表3。
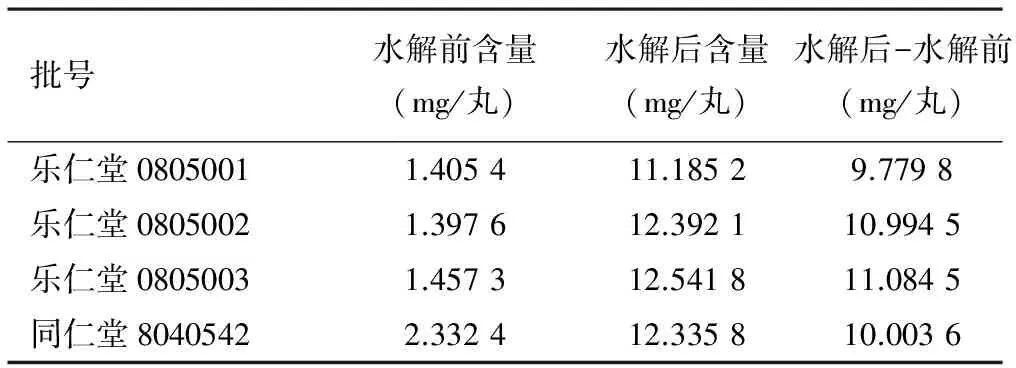
表3 4批样品含量测定结果
4 讨论
4.1 麝香酮采用二硝基苯肼乙醇试液显色显黄色,但是薄层板背景颜色也同样显黄色(见图3),影响斑点检视效果。试验中采用先加显色剂并加温反应后,再点样、展开的方式,结果消除了薄层板背景干扰现象,结果满意。
4.2 五倍子中主含五倍子鞣质,含量为60%~70%,五倍子鞣质是由葡萄糖与没食子酸结合所成,同时亦含游离的没食子酸,含量占2%~4%。五倍子鞣质具有敛肺、涩肠、止血、解毒功效。因此,起草标准采用高效液相色谱法,分别对样品中游离的没食子酸与总没食子酸进行了测定。
4.3 样品水解方式的考查 取同一50%甲醇加热回流提取后的样品溶液,其中两份样品溶液减压蒸干,其余两份样品保留,分别加入盐酸溶液(36→100)20 ml,置沸水浴中加热回流2 h,放冷,转移至50 ml量瓶中,用50%甲醇稀释至刻度,摇匀,取续滤液,即得。结果减压蒸干的样品水解后,样品中总没食子酸含量较高,因此样品应减压蒸干后再加盐酸水解。原因分析:甲醇的沸点为64.5 ℃,如果样品溶液中含有甲醇,在水浴中加热时会首先沸腾、蒸发、冷凝,导致样品溶液实际水解温度降低,引起样品中的没食子酸含量低于实际结果,见表4。

表4 水解方式的考查(n=2)
4.4 色谱条件的选择 《中国药典》2005年版一部五倍子含量测定采用的流动相为甲醇-0.1%磷酸(15∶85),经试验,此流动相没食子酸色谱峰出峰较快,因此将流动相比例调整为甲醇-0.1%磷酸(10∶90)[4,5]。
1 高咏莉.木香理气片的质量标准研究[J].天津药学,2007,19(4):18-19
2 周军,张晶,金兆祥,等.济坤丸质量标准研究[J].天津药学,2010,22(1):28-30
3 苏海萍.咽炎糖浆的制备及质量控制[J].天津药学,2004,16(3):14-15
4 夏清,刘圆,李莹,等.RP-HPLC测定鬼针草属药材中的没食子酸[J].华西药学杂志,2009,24(3):308-310
5 中国药典[S].一部.2005:45-46
2016-01-26
R927
A
1006-5687(2016)02-0004-04
