超高效液相色谱-质谱联用法测定草莓中鞣花酸
2017-01-06杨军军张莹莹张开春
杨 媛,石 磊,杨军军,张莹莹,张开春,3*
(1.北京市农林科学院 林业果树研究所,北京 100093;2.农业部华北地区园艺作物生物学与种质创制重点实验室,北京 100097;3.北京市落叶果树工程技术研究中心,北京 100093)
超高效液相色谱-质谱联用法测定草莓中鞣花酸
杨 媛1,2,石 磊1,杨军军1,张莹莹1,张开春1,3*
(1.北京市农林科学院 林业果树研究所,北京 100093;2.农业部华北地区园艺作物生物学与种质创制重点实验室,北京 100097;3.北京市落叶果树工程技术研究中心,北京 100093)
建立了测定草莓中鞣花酸含量(包括游离鞣花酸和总鞣花酸)的超高效液相色谱-质谱联用(UPLC-MS/MS)分析方法。草莓中游离鞣花酸在酸性条件下用甲醇提取,经C18分散固相萃取净化后可直接测定;总鞣花酸经酸性水解呈游离态再经分散固相萃取净化后进行测定。净化液经C18色谱柱分离,以甲醇和0.5%甲酸水为流动相进行梯度洗脱,电喷雾负离子(ESI-)模式电离,超高效液相色谱-质谱法(UPLC-MS/MS)测定,外标法定量。结果表明:在10~500 ng/mL浓度范围内鞣花酸的线性关系良好,相关系数为0.998 1,游离鞣花酸的定量下限为0.5 mg/kg,总鞣花酸的定量下限为5.0 mg/kg。在低、中、高3个加标浓度的回收实验中,游离鞣花酸的加标回收率为86.7%~113.6%,相对标准偏差均小于10%。该方法操作简单,灵敏度高,准确性好,适用于草莓中鞣花酸的测定。
超高效液相色谱-质谱联用法(UPLC-MS/MS);游离鞣花酸;总鞣花酸;草莓
近年来,鞣花酸因其突出的抗氧化效果备受关注。有报道表明,鞣花酸广泛存在于山莓[1]、草莓[2]、蓝莓[3]、红醋栗[4]、石榴[5]、胡桃[6]等水果和坚果中。鞣花酸具有重要的抗菌[7]、抗炎[8]、抗氧化[9-12]、促进伤口愈合[4]、心肌保护[13]、镇痛[14]、肿瘤抑制[1,15]等功能,对癫痫病[16]、糖尿病及其并发症[17]、动脉硬化[18]、癌症[19-21]等疾病的预防和治疗效果备受关注,鞣花酸具有比维生素E更强的抗氧化胁迫作用[22]。
鞣花酸存在3种形式[1]:即与糖结合成醚的鞣花单宁,鞣花酸糖苷,以及少量的游离鞣花酸。目前,鞣花酸含量的分析方法有高效液相色谱法[23-24]、反滴定法[25]、紫外分光光度法[4,26]、薄层层析法[27-28]、高效毛细管电泳法[29]等,但国内鲜有关于超高效液相色谱-质谱联用法(UPLC-MS/MS)检测鞣花酸的报道。在鞣花酸测定的多种方法中,反滴定法的操作比较繁琐、费时费力且溶剂用量大;分光光度法前处理采用萃取法比较繁琐,且杂质干扰影响测定结果;薄层层析法主要用于定性分析;高效毛细管电泳法前处理复杂、耗时长、溶剂用量大;高效液相色谱法是目前效率较高、定性定量准确性好的一种检测方法。而相比之下,超高效液相色谱-质谱联用法具有效率更高,溶剂试剂用量更少,定性、定量更准确的优势。
本研究针对富含鞣花酸的代表性水果草莓[30],建立了分散固相萃取净化、超高效液相色谱-质谱联用法测定草莓中游离鞣花酸和总鞣花酸,该方法操作简便,准确性高,可用于其他水果中游离鞣花酸和总鞣花酸含量的测定。
1 实验部分
1.1 仪器与试剂
Waters Xevo TQ-S(Acquity UPLC,ZspraYTM,ESI/APCI/ESCi®,Waters公司);乙腈、甲醇(色谱纯,Merck公司),三氟乙酸(分析纯,北京北化精细化学品有限责任公司),鞣花酸(纯度98.5%,Dr.Ehrenstorferg公司),C18填料(Dikma公司)。除非另有说明,其他所用试剂均为分析纯试剂;实验用水为GB/T6682规定的一级水。
1.2 样品前处理
1.2.1 游离鞣花酸 准确称取1.00 g草莓匀浆样品置于50 mL具塞刻度离心管中,加入甲醇-0.1%三氟乙酸水(80∶20)提取液定容至50 mL,涡旋混匀3 min,10 000 r/min离心5 min。取2 mL上清液加入20 mg C18粉末,涡旋振荡3 min,5 000 r/min离心5 min。取上清液过0.22 μm有机滤膜,滤液供UPLC-MS/MS测定。
1.2.2 总鞣花酸 称取1.00 g 草莓匀浆样品置于50 mL刻度离心管中,加入2 mol/L三氟乙酸溶液定容至50 mL,涡旋振荡3 min,置于90 ℃水浴中加热2 h。冷却后10 000 r/min离心5 min,取1 mL上清液置于10 mL刻度离心管中,加入甲醇定容至10 mL,涡旋振荡2 min。取2 mL上清液加入20 mg C18粉末,涡旋振荡2 min,10 000 r/min离心5 min。取1.0 mL清液,过0.22 μm有机相滤膜,滤液供UPLC-MS/MS测定。
1.3 标准溶液配制
鞣花酸标准储备液浓度为1.0 mg/mL的乙腈溶液,避光存储于4 ℃冰箱,用甲醇逐级稀释为500,250,100,50,25,10 ng/mL的标准工作溶液,使用前需新鲜配制。
1.4 检测条件
1.4.1 液相色谱条件 色谱柱:Acquity UPLC BEH®C18(2.1×50 mm,1.7 μm);柱温:30 ℃;流动相:A为甲醇,B为0.5%甲酸水溶液;流速:0.2 mL/min;进样量:1.0 μL。梯度洗脱程序:0~2.5 min,90%B;2.5~3 min,90%~10%B;3~5 min,10%B;5~5.5 min,10%~90%B;5.5~8 min,90%B。
1.4.2 质谱条件 扫描模式:电喷雾负离子模式ESI(-);毛细管电压:2 500 V;干燥气流量:氮气 500 L/H;去溶剂化温度:350 ℃;雾化器压力:7.0 bar;碰撞气:氩气;监测方式:多反应监测(MRM);定量离子对300.97>144.70,锥孔电压82 V,碰撞电压38 V;定性离子对300.97>228.71,锥孔电压82 V,碰撞电压28 V。
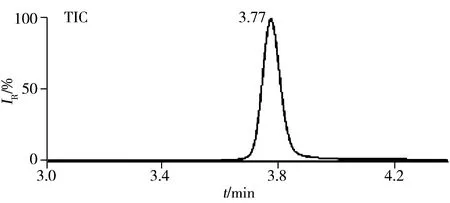
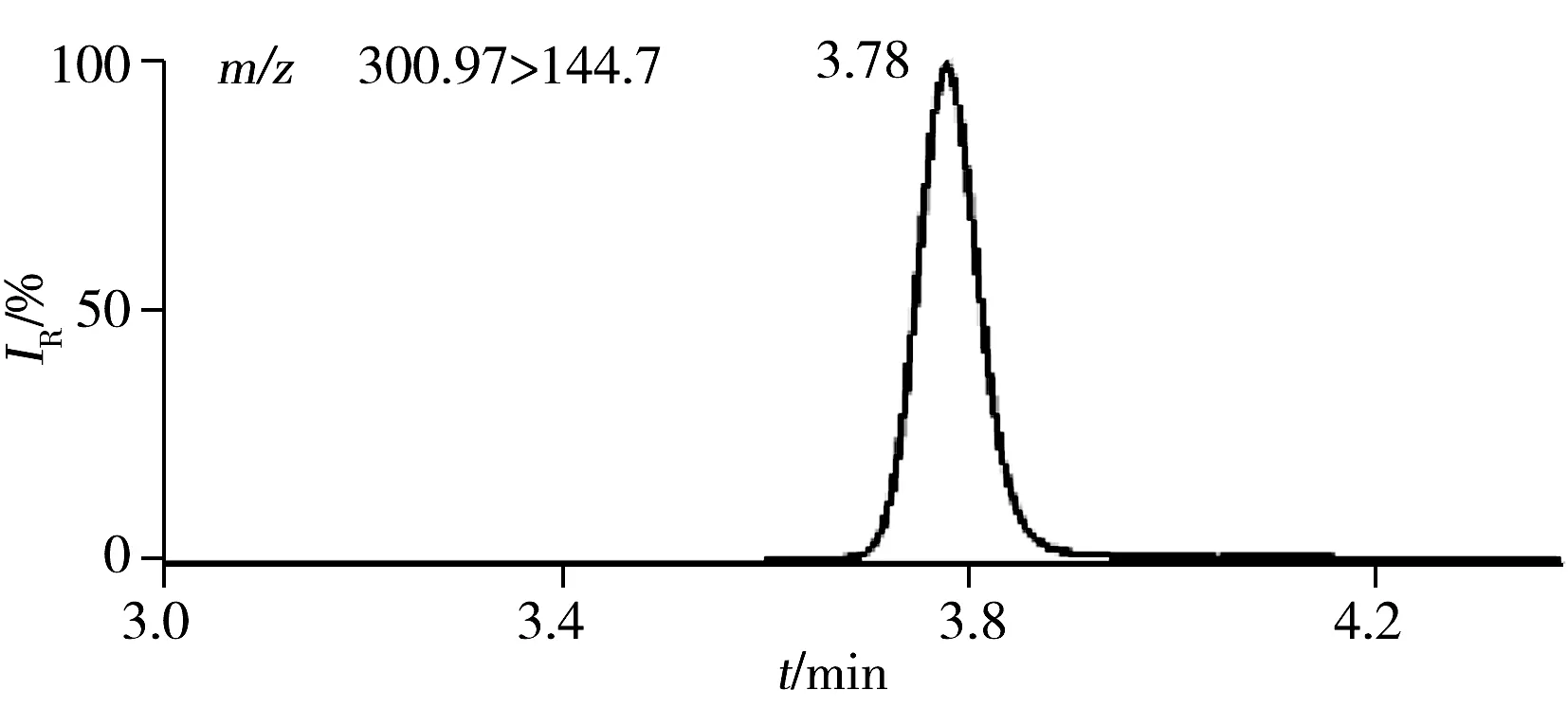
图1 50 ng/mL鞣花酸的典型色谱质谱图Fig.1 MRM chromatograms of 50 ng/mL ellagic acid

图2 鞣花酸的二级质谱图Fig.2 MRM spectrum of ellagic acid collision energy:38 eV
2 结果与讨论
2.1 质谱条件的优化
本研究采用UPLC-MS/MS技术进行检测,由于二级质谱的多反应离子监测模式与一级质谱的单离子监测方式相比专属性更高,抗背景干扰能力更强,定量的准确性更好,检出限更低,因此本文以多反应离子监测模式检测鞣花酸;扫描方式的选择主要依据物质本身的结构特点,鞣花酸作为有机酸易失去1个质子带负电荷,理论上鞣花酸可采用负离子进行扫描。研究证实,在含有甲酸的流动相系统中,采用负离子方式扫描时鞣花酸的响应较高,峰形较尖锐,因此,本研究选择多反应离子监测模式负离子扫描方式检测鞣花酸(见图1)。
2.2 裂分途径
图2为碰撞电压为38 eV条件下,鞣花酸的二级质谱图。根据鞣花酸的分子结构特点和碎片质荷比,推测鞣花酸在该质谱条件下的裂分途径如图3所示。由图3可见,鞣花酸(化合物(1))是含有4个羟基的对称双内酯,在负离子模式下,容易失去1个氢离子带上负电荷而形成较稳定的分子离子即(2),该离子由于分子的良好对称性和苯环的π键共轭作用而能稳定存在。鞣花酸有3个响应较高的碎片离子,其中,m/z=283.82是分子离子丢失水分子的产物即(3)。m/z=228.71是(3)丢失两分子CO的产物即(4);m/z=144.70是(4)进一步丢失3分子CO的产物即(5)。
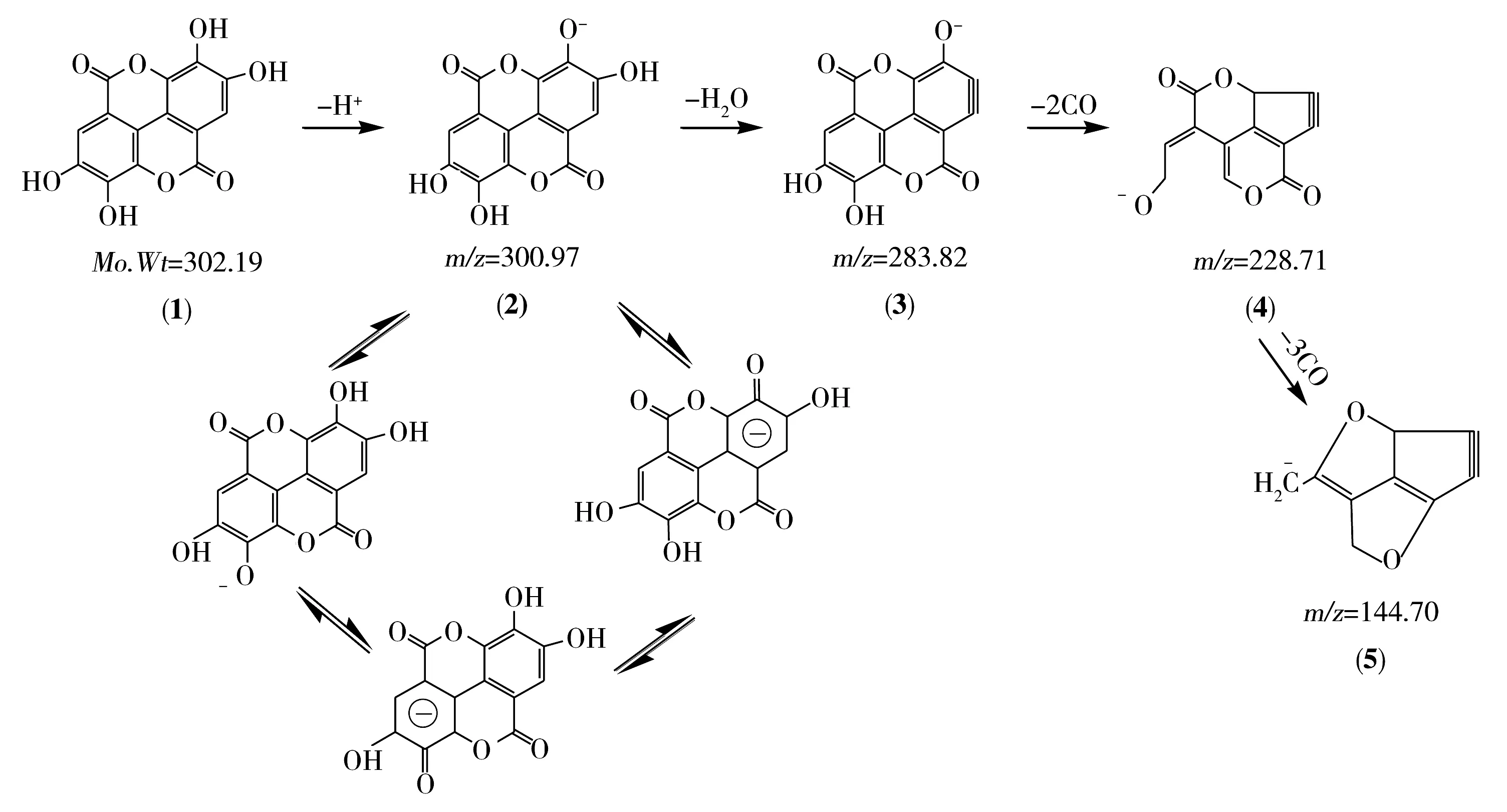
图3 负离子模式下鞣花酸的质谱裂分示意图Fig.3 Tentative assignment of fragmentation of ellagic acid under negative ion mode
2.3 前处理条件的选择
2.3.1 游离鞣花酸提取溶剂的选择 目前国内报道的高效液相色谱法中,鞣花酸的提取多采用丙酮溶液浸泡6~8 h后超声或回流提取,其操作繁琐、耗时长,且提取后未经净化直接使用高效液相色谱仪检测,易对色谱柱及检测器造成污染。
鞣花酸属于酸性物质,本实验比较了乙腈、甲醇、乙腈-水(80∶20)、甲醇-水(80∶20)、乙腈-0.1%三氟乙酸水溶液(80∶20)、甲醇-0.1%三氟乙酸水溶液(80∶20) 6种提取液对“章姬”样品中添加5.0 mg/kg鞣花酸的提取效果。结果表明,鞣花酸在后2种溶剂中的提取回收率均大于90%,而在前4种溶剂中的提取回收率均不大于60%,表明后2种溶剂的提取效果较好。但样品经乙腈-0.1%三氟乙酸水溶液(80∶20)提取,C18净化后,仍含有较多杂质;而经甲醇-0.1%三氟乙酸水溶液(80∶20)提取,C18净化后可有效去除杂质。因此,实验选择甲醇-0.1%三氟乙酸水溶液(80∶20)作为样品的最佳提取溶剂。
2.3.2 总鞣花酸水解条件的优化 用“章姬”样品比较了总鞣花酸在酸性条件(2 mol/L三氟乙酸水溶液)和碱性条件(10 mol/L氢氧化钠溶液)的水解效果。结果表明,两种水解方式下总鞣花酸的水解率相当。但碱性水解后,反应溶液还需进行pH值调节和萃取分液等步骤后,才能进行稀释、上机测定;而酸性水解后,反应液可直接进行稀释、上机测定。因此,实验选择步骤更为简化的酸性方式将总鞣花酸水解为游离鞣花酸。
2.4 线性范围与定量下限
在优化色谱条件下,测定了一系列浓度(500,200,100,50,25,10 ng/mL)的鞣花酸标准溶液。分别以定量离子峰面积(Y)对质量浓度(X,ng/mL)进行回归方程拟合。结果表明,在10~500 ng/mL浓度范围内,鞣花酸的线性关系良好,线性方程为Y=97.67X+2 396(r=0.998 1 )。该方法的最低定量浓度为10 ng/mL,根据前处理条件,鞣花酸的定量下限为0.5 mg/kg,总鞣花酸(以游离鞣花酸计算)的定量下限为5.0 mg/kg。
2.5 加标回收率及相对标准偏差
选取“红颜”、“章姬”、“甜查理”3种北京主栽草莓品种为游离鞣花酸加标回收实验的基质,分别添加0.5,5.0,10 mg/kg游离鞣花酸,每个浓度设3个平行。实验结果表明,3种代表基质中游离鞣花酸的加标回收率为86.7%~113.6%,相对标准偏差(RSD)为2.1%~9.5%(见表1)。表明方法的精密度和准确度较好,能满足检测要求。
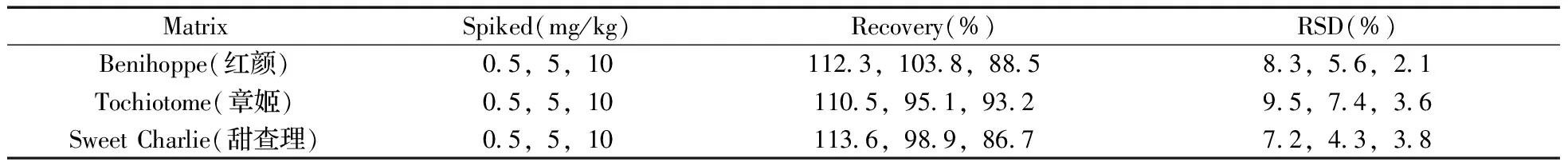
表1 草莓中游离鞣花酸的添加回收试验结果(n=3)
2.6 重现性试验
平行称取同一“章姬”匀浆样品6份,分别置于50 mL具塞刻度离心管中,加入甲醇-0.1%三氟乙酸水(80∶20)提取液定容至50 mL,涡旋混匀3 min,10 000 r/min离心5 min。取2 mL上清液加入20 mg C18粉末,涡旋振荡3 min,5 000 r/min离心5 min。取上清液过0.22 μm有机滤膜,滤液供UPLC-MS/MS测定。结果表明,该样品的游离鞣花酸含量为2.30 mg/kg,RSD为2.4%,具有良好的重复性。
平行称取同一“章姬”匀浆样品6份,分别置于50 mL刻度离心管中,加入2 mol/L三氟乙酸溶液定容至50 mL,涡旋振荡3 min,置于90 ℃水浴中加热2 h。冷却后10 000 r/min离心5 min,取1 mL上清液置于10 mL刻度离心管中,加入甲醇定容至10 mL,涡旋振荡2 min。取2 mL上清液加入20mg C18粉末,涡旋振荡2 min,10 000 r/min离心5 min。取1.0 mL清液,过0.22 μm有机相滤膜,滤液供UPLC-MS/MS测定。结果表明,该样品的总鞣花酸含量为26.54 mg/kg,RSD为1.3%,具有良好的重复性。
2.7 实际样品分析
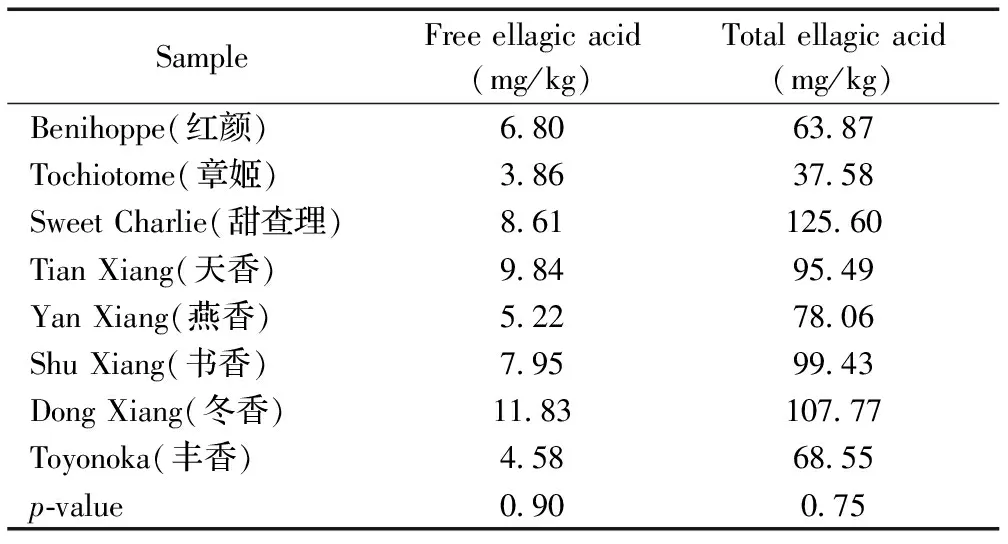
采用本方法对市售8种实际草莓样品进行分析,结果表明,不同品种草莓中所含的游离鞣花酸含量差别较小,但总鞣花酸含量有所差别(见表2),但无论是游离鞣花酸还是总鞣花酸,几种草莓间均不具有显著性差异,即相互间未达到p<0.05显著水平。
表2 8种草莓中鞣花酸含量的测定结果(n=2)
Table 2 Content of ellagica acid in eight strawberry samples (n=2)
SampleFreeellagicacid(mg/kg)Totalellagicacid(mg/kg)Benihoppe(红颜)6806387Tochiotome(章姬)3863758SweetCharlie(甜查理)86112560TianXiang(天香)9849549YanXiang(燕香)5227806ShuXiang(书香)7959943DongXiang(冬香)118310777Toyonoka(丰香)4586855p⁃value090075
3 结 论
本文建立了超高效液相色谱-质谱联用法测定草莓中鞣花酸(包括游离鞣花酸和总鞣花酸),游离鞣花酸在酸性条件下用甲醇提取,经C18分散固相萃取净化后可直接测定;总鞣花酸经酸性水解成游离态后,再进行分散固相萃取净化后,上机测定。方法前处理简单快捷,测定结果稳定、灵敏度高、准确性好,适合于草莓中鞣花酸的测定,同时也可为其他水果中鞣花酸的测定提供参考。
[1] Flores G,Ruiz del Castillo M L.J.FoodCompos.Anal.,2015,39:55-61.
[2] Ghorbanzadeh B,Mansouri M T,Hemmati A A,Naghizadeh B,Mard S A,Rezaie A.Pharmacol.Biochem.Behav.,2014,126:116-121.
[3] Priyadarsini K I,Khopde S M,Kumar S S,Mohan H.J.Agric.FoodChem.,2002,50:2200-2206.
[4] Gopalakrishnan L,Raman N R,Sethuraman S,Krishnan U M.Carbohydr.Polym.,2014,111:215-221.
[5] Buenrostro-Figueroa J,Huerta-Ochoab S,Prado-Barragánb A,Ascacio-Valdésa J,Sepúlvedaa L,Rodrígueza R,Aguilera-Carbóc A,Aguilar C N.ProcessBiochem.,2014,49:1595-1600.
[6] Mansouri M T,Naghizadeh B,Ghorbanzadeh B.Pharmacol.Biochem.Behav.,2014,120:43-49.
[7] Farbood Y,Sarkaki A,Dianat M,Khodadadi A,Haddad M K,Mashhadizadeh S.LifeSci.,2015,124:120-127.
[8] El-Shitany N A,El-Bastawissy E A,El-desoky K.Int.Immunopharmacol.,2014,19:290-299.
[9] Lee W J,Ou H C,Hsu W C,Chou M M,Tseng J J,Hsu S L,Tsai K L,Sheu W H.J.Vasc.Surg.,2010,52(5):1290-1300.
[10] Qiu Z,Zhou B,Jin L,Yu H,Liu L,Liu Y,Qin C,Xie S,Zhu F.FoodChem.Toxicol.,2013,59:428-437.
[11] Vattem D A,Shetty K.ProcessBiochem.,2003,39(3):367-379.
[12] Fukuda T,Ito H,Yoshida T.Phytochemistry,2003,63(7):795-801.
[13] Warpe V S,Mali V R,Arulmozhi S,Bodhankar S L,Mahadik K R.J.AcuteMed.,2015,5:1-8.
[14] Mansouri M T,Naghizadeh B,Ghorbanzadeh B.Pharmacol.Rep.,2015,67:473-477.
[15] Losso J N,Bansode R R,Trappey A,Bawadi H A,Truax R.J.Nutr.Biochem.,2004,15:672-678.
[16] Dhingra D,Jangra A.J.Funct.Foods,2014,10:364-369.
[17] Akileshwari C,Raghu G,Muthenna P,Mueller N H,Suryanaryana P,Petrash J M,Reddy G B.J.Funct.Foods,2014,6:374-383.
[18] Elhemely M A,Omar H A,Ain-Shoka A A,Abd El-Latif H A,Abo-Youssef A M,El Sherbiny G A.Beni-suefUniv.J.BasicAppl.Sci.,2014,3:239-246.
[19] Munagala R,Aqil F,Vadhanam M V,Gupta R C.CancerLett.,2013,339:175-184.
[20] Mertens-Talcott S U,Talcott S T,Percival S S.J.Nutr.,2003,133:2669-2674.
[21] Notka F,Meier G,Wagner R.AntiviralRes.,2004,64:93-102.
[22] Hassonna E A,Walter A C,Alsharif N Z,Stohs S J.Toxicology,1997,124:27-37.
[23] Nankar R P,Doble M.J.Funct.Foods,2015,15:1-10.
[24] Yang Y,Shi L,Zhang J,Li W S,Zhang K C,Feng X Y.NorthernHorticulture(杨媛,石磊,张佳,李文生,张开春,冯晓元.北方园艺),2012,15:25-27.
[25] Zhang Q.J.NorthwestUniv.:Nat.Sci.Ed.(张强.西北大学学报:自然科学版),1997,27(4):313-315.
[26] Chen J H,Wu D M,Wang Y M,Wu Z S.BiomassChem.Eng.(陈笳鸿,吴冬梅,汪咏梅,吴在嵩.生物质化学工程),2007,41(3):18-20.
[27] Lin T Y,Vine R P.J.FoodSci.,1990,55(6):1607-1609.
[28] Avachat A M,Patel V G.SaudiPharm.J.,2015,23:276-289.
[29] Zhou B H,Wu Z H,Li X J,Liu C,Zhang J.Chin.Pharm.(周本宏,吴振华,李小军,刘春,张杰.中国药房),2005,16(24):1893-1894.
[30] Zheng Y L,Wang S Y,Wang C Y,Zheng W.LWT-FoodSci.Technol.,2007,40:49-57.
Determination of Ellagic Acid in Strawberry by UPLC-MS/MS
YANG Yuan1,2,SHI Lei1,YANG Jun-jun1,ZHANG Ying-ying1,ZHANG Kai-chun1,3*
(1.Institute of Forestry and Pomology,Beijing Academy of Agriculture and Forestry Sciences,Beijing 100093,China;2.Key Laboratory of Biology and Genetic Improvement of Horticultural Crops(North China),Ministry of Agriculture,China,Beijing 100097,China;3.Beijing Engineering Research Center for Deciduous Fruit Trees,Beijing 100093,China)
A method was developed for the determination of ellagic acid(free ellagic acid and total ellagic acid) in strawberry by ultra performance liquid chromatography-tandem mass spectrometry(UPLC-MS/MS).In this work,the free ellagic acid was extracted from strawberry with 0.1%trifloroacetric acid-methane,purified by C18dispersive solid-phase extraction,and determined by UPLC-MS/MS.Total ellagic acid was hydrolyzed to free ellagic acid with 2 mol/L trifloroacetric acid aqueous,then purified with C18dispersive solid-phase extraction column,and determined by UPLC-MS/MS.Sample solution was separated on a C18column by gradient elution using methanol-0.5%formic acid aqueous as mobile phase.The linear range of 10-500 ng/mL for ellagic acid was obtained,with correlation coefficient of 0.998 1.The limit of quantitation was 0.5 mg/kg for free ellagic acid and 5.0 mg/kg for total ellagic acid.The recoveries at low,medium and high spiked levels ranged from 86.7%to 113.6%with relative standard deviations(RSD) not more than 10%.The results indicated that this approach was simple,sensitive and accurate,and was applicable for the determination of content of ellagic acid in strawberry.
ultra performance liquid chromatography-tandem mass spectrometry(UPLC-MS/MS);free ellagic acid;total ellagic acid;strawberry
2016-06-15;
2016-07-25
科技创新能力建设专项(KJCX20140302,KJCX20150602)
10.3969/j.issn.1004-4957.2016.12.013
O656.63;S816.7
A
1004-4957(2016)12-1591-05
*通讯作者:张开春,博士,研究员,研究方向:果树资源育种及果品质量安全,Tel:010-82596007,E-mail:zkc8848@126.com
