离子色谱-柱后衍生-紫外可见检测法测定地下水中痕量六价铬
2016-12-30周健楠杨懂艳丁萌萌刘保献
徐 硕 周健楠 杨懂艳 丁萌萌 常 淼 刘保献
(北京市环境保护监测中心,北京 100048)
离子色谱-柱后衍生-紫外可见检测法测定地下水中痕量六价铬
徐 硕1周健楠 杨懂艳 丁萌萌 常 淼 刘保献
(北京市环境保护监测中心,北京 100048)
建立了离子色谱-柱后衍生-紫外可见检测法直接测定地下水中痕量六价铬的方法,确定了方法的性能指标,开展了干扰实验的研究。研究发现,本方法可在8分钟内完成分析,六价铬峰型尖锐,方法标准曲线线性范围0.05~5.00 μg/L,相关系数≥0.9999。方法检出限较低(0.008 μg/L),精密度和准确度较高,3种地下水实际样品的加标回收率为94%~106%,RSD为0.6%~4.0%(n=8)。高浓度SO42-(500~3000 mg/L)对六价铬测定无干扰,而Cl-(>500 mg/L)可在一定程度上干扰六价铬峰形。本方法耗时短、选择性强、灵敏度高、前处理简单、干扰较少,相对常规方法能够更好地满足地下水中六价铬测定的实际需要。
离子色谱 柱后衍生 地下水 六价铬
六价铬Cr(Ⅵ)在天然水体中具有很好的溶解性,多以CrO42-和HCrO4-的形式存在。六价铬对人类、动物及水生生物均有毒害作用,其半致死量ED50是三价铬的100倍,被国际癌症研究署(IARC)列为一级致癌物[1]。我国《地下水质量标准》(GB/T 14848-93)中明确规定六价铬的I类限值为0.005 mg/L[2]。
目前,地下水中六价铬的测定方法主要有二苯碳酰二肼分光光度法、离子色谱法、流动注射分光光度法、原子吸收光谱法、原子荧光光谱法以及ICP-AES等,其中国家标准(GB 7467-87)[3]采用二苯碳酰二肼分光光度法,最低检出浓度0.004 mg/L,虽略低于I类限值,但无法满足地下水中痕量六价铬的定量检测,且该方法易受样品浊度、色度及干扰离子的影响,容易出现假阳性或假阴性结果、重现性差。
离子色谱法可有效排除样品基体干扰,色谱柱的分离作用使得六价铬单独进入检测器。与常见的电导检测器相比,柱后衍生紫外可见检测可以特异性地检出六价铬,而对其他阴离子无响应,因此在测定时干扰更少、灵敏度更高。本实验采用离子色谱法直接测定地下水中的六价铬,水样中以铬酸盐(CrO42-)形式存在的六价铬经阴离子色谱柱与样品基体分离,由在线柱后衍生,流入紫外可见检测器,于530 nm处检测,考察了方法的准确性、重现性和实用性,获得了方法的性能指标。
1 实验部分
1.1 试剂与仪器
Dionex ICS-3000型离子色谱仪(美国赛默飞世尔科技公司),配有柱后衍生装置、Dionex Ultimate 3000可变波长紫外可见检测器;Chromeleon 6.8色谱工作站; 0.45μm微孔滤膜过滤头(水系)。
硫酸铵(分析纯,国药集团化学试剂有限公司);氨水(优级纯,国药集团化学试剂有限公司);二苯碳酰二肼(分析纯,国药集团化学试剂有限公司);甲醇(HPLC级,TEDIA);浓硫酸(优级纯,北京化工厂);六价铬标准溶液(100μg/mL,国家标准物质研究中心);实验用水均为新制备的二次去离子水,电阻率>18.2 MΩ·cm。
1.2 色谱条件
色谱柱:IonPac AS7分离柱(4 mm×250 mm),IonPac NG1保护柱(4 mm×35 mm);淋洗液:250 mmol/L 硫酸铵-100 mmol/L 氨水,1.0 mL/min;柱后衍生剂:2 mmol/L 二苯碳酰二肼(DPC)-0.5 mol/L 硫酸-10%(V/V)甲醇,0.30 mL/min;进样体积:900 μL;检测波长:530 nm。
1.3 标准曲线绘制
取六价铬标准溶液,配制成0.05μg/L、0.20 μg/L、0.50 μg/L、1.00 μg/L、2.00 μg/L、5.00μg/L共6个浓度的标准系列,按上述色谱条件进行测定。以六价铬浓度为横坐标,仪器响应值(峰面积)为纵坐标绘制标准曲线。
1.4 样品制备
地下水样品采集后于0~4℃运输或保存,24 h内测定,测定前使用0.45 μm滤膜过滤,舍去1~2mL初滤液,收集续滤液,置于自动进样器样品瓶中,待测。
2 结果与讨论
2.1 六价铬标准谱图
依照1.2节所述色谱条件,测定1.0μg/L的六价铬标准溶液,标准谱图如图1所示。方法可在8 min内完成分析,六价铬保留时间在5.9 min附近,峰形尖锐,周围无杂峰,方法的色谱条件可满足六价铬的检测需求。
2.2 方法线性及检出限
根据1.3所述标准曲线绘制方法,开展方法标准曲线线性实验,结果表明六价铬质量浓度在0.05~5.00μg/L时,峰面积Y与六价铬浓度X(μg/L)线性关系良好,线性回归方程为Y=0.347X,相关系数r=0.9999。标准曲线如图2所示。
根据《环境监测分析方法标准制修订技术导致》(HJ168-2010)[4],配制浓度为0.05 μg/L的六价铬标准溶液,按上述色谱条件平行测定7次,结果见表1。经计算,本方法直接测定六价铬的方法检出限为0.008 μg/L,测定下限为0.032 μg/L,比国标方法GB 7467-87的最低检出浓度(0.004 mg/L)低两个数量级,可很好地满足《地下水质量标准》(GB/T 14848-93)[2]中六价铬的测定需求。

表1 方法检出限测定结果
2.3 精密度和准确度
为考察实验室内空白加标样品的精密度和准确度,使用纯水分别配制0.04μg/L、1.00 μg/L、5.00 μg/L的六价铬标准溶液,平行测定6次,RSD值在0.7%~3.7%之间(见表2);同时将浓度为500 μg/L和54.9 μg/L的有证标准物质稀释100倍,平行测定6次,RE值分别为0.2%和2.6%(见表3)。

表2 方法精密度测定结果

表3 方法准确度测定结果
为考察本方法测定不同种类实际地下水的精密度和准确度,选取3种地下水井(平原区域地下水井、工业区水井、水源井)采集水样。样品连续进样测定8次,RSD为0.6%~4.0%,样品加标后测得回收率为94%~106%(见表4)。由此可见,采用离子色谱-柱后衍生-紫外可见检测法直接测定地下水中痕量六价铬,方法精密度和准确度较高,可满足实际检测需求。
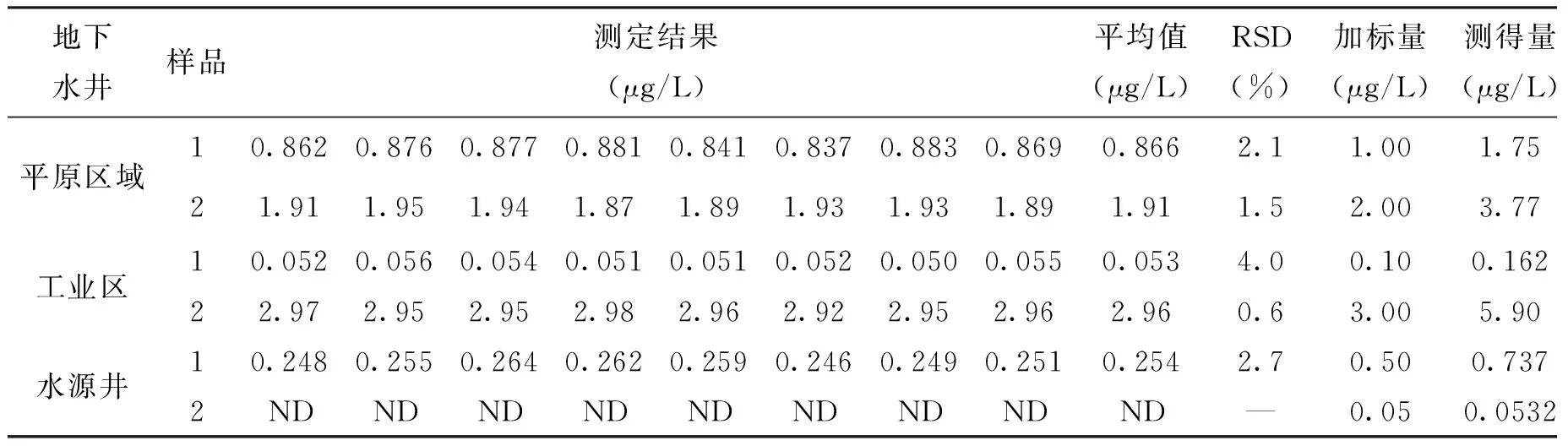
表4 实际样品测定结果及其加标回收率
2.4 方法干扰试验
根据《水质六价铬的测定二苯碳酰二肼分光光度法》(GB 7467-87)[3],浊度、色度及干扰离子(铁、钼、汞、钒等)都会影响六价铬的显色。本实验法利用离子色谱柱将六价铬由样品基体中分离出来,有效排除了上述因素的干扰。
高浓度阴离子(如Cl-、SO42-)可导致色谱柱过载,表现为加标回收率降低和色谱峰拖尾。实验室内对北京市地下水样品的测定表明Cl-、SO42-的浓度一般为0~300 mg/L。查阅资料可知普通地下水Cl-、SO42-的浓度一般在500 mg/L以下[5,6];但部分污灌区地下水、矿区地下水、盐化地下水中Cl-、SO42-浓度接近3000mg/L[7]。不同浓度阴离子(Cl-、SO42-)对六价铬(1.00 μg/L)测定的影响如图3所示。可见,随着Cl-浓度的增加,六价铬峰高逐渐降低,但峰面积不变,即出现峰形变宽变矮的现象。SO42-浓度的增加并未对六价铬峰形造成显著影响,峰面积和峰高变化不大。因此,地下水中六价铬的测定可能会受到高浓度Cl-(>500 mg/L)的干扰,实际测定时应考察六价铬峰形及加标回收率。
3 结论
地下水中六价铬浓度一般为10-9(μg/L)级别甚至更低。在地下水的实际测定中,目前普遍采用的二苯碳酰二肼分光光度法对大部分水样的测定结果均是未检出。本实验所用的离子色谱-柱后衍生-紫外可见检测法,检出限低至0.008 μg/L,可检测出地下水中六价铬的实际浓度,为相关的痕量六价铬研究提供了可靠的手段。在已配备有离子色谱仪的实验室中,此方法可快速建立,简单易行。对于以水质评判为目的的六价铬监测,离子色谱-柱后衍生-紫外可见检测法相对于二苯碳酰二肼分光光度法的优势在于能够连续自动进样分析,且不受样品基体干扰,十分适用于大量水样或干扰物质较多的不清洁水样的定量检测。但对于少量的清洁水样,由于离子色谱法试剂需现用现配,采用二苯碳酰二肼法更为合适。
水中六价铬的其它测定方法中,流动注射分光光度法需要复杂的预处理来排除浊度、色度等样品基体的干扰[8-10];原子吸收光谱法、原子荧光光谱法、电感耦合等离子光谱及质谱法只能测定总铬,或需要繁琐的离子分离步骤才能测定六价铬,影响分析速度[11];离子色谱分离-电导检测法的检出限为5 μg/L[12],改进后的毛细管离子色谱分离-电导检测法检出限低至1.0 ng/L,但分析时间长达41 min,同时其样品色谱图中杂峰较多,六价铬响应信号并不突出[13]。
本实验选用的离子色谱-柱后衍生-紫外可见检测法,可将样品过滤后直接进样,分析耗时8 min;六价铬峰形尖锐,周围无杂峰;经标准样品及实际样品测定,方法具有非常好的准确性及重现性。因此,本方法在水中痕量六价铬的检测方面具有良好的应用前景。
[1] IARC.IARC monographs on the evaluation of carcino-genic risks to humans volume 49 chromium, nickel and welding[R].Geneva:World Health Organization,1997:17-33.
[2] GB/T 14848-93,地下水质量标准[S].
[3] GB 7467-87,水质六价铬的测定二苯碳酰二肼分光光度法[S].
[4] HJ 168-2010,环境监测分析方法标准制修订技术导则[S].
[5] 刘春,谭利敏,尹国勋,范俊玲.焦作市某污灌区地下水无机氯化物污染原因初探[J].江苏环境科技,2006,19(z2):124-126.
[6] 倪传钧.淮南潘集矿区地下水水化学特征分析[J].安徽科技,2006(2):44-46.
[7] 李彬,史海滨,张建国,李祯.节水改造前后内蒙古河套灌区地下水水化学特征[J].农业工程学报,2014,30(21):99-110.
[8] ISO23913:2006,Water quality — Determination of chromium(Ⅵ)— Method using flow analysis (FIA and CFA)and spectrometric detection [S].
[9] 林志鹏,刘海术,杨芳.流动注射分光光度法测定水中六价铬的研究[J].干旱环境监测,2014,28(2):66-69, 93.
[10] 樊静,陈亚红,冯素玲.流动注射在线分离预浓集分光光度法测定环境水样中的痕量铬(Ⅵ)[J].分析试验室,2004,23(1):70-72.
[11] 丁红红,郑明凯.饮用水中六价铬测定方法进展[J].广州化工,2012,40(17):39-40, 82.
[12] 严利民,胡文武.离子色谱法测定水中六价铬[J].中国热带医学,2007,7(1):87-88.
[13] 胡忠阳,汪琼,叶明立,梁立娜,何世伟,朱岩.离子色谱法测定饮用水中的六价铬[J].中国无机分析化学,2012,02(z1):1-2.
Determination of trace Cr(Ⅵ) in groundwater by ion chromatography(IC) with post-column derivatization and UV/Visible spectrometer.
Xu Shuo, Zhou Jiannan, Yang Dongyan, Ding Mengmeng, Chang Miao,Liu Baoxian
(BeijingMunicipalEnvironmentalMonitoringCenter,Beijing100048,China)
The method was proved to be effective in Cr(Ⅵ) determination, and the analysis was accomplished in 8 minutes with a favorable peak profile. The correlation coefficient was >0.9999 for Cr(Ⅵ) in the range of 0.05-5 μg/L, and the detection limit was as low as 0.008 μg/L. The high accuracy and precision were proved by the satisfactory recovery rates (94%-106%) and RSDs (0.6%-4.0%) for 3 different kinds of actual groundwater samples. The high concentration of sulfate had no influence in Cr(Ⅵ) detection while the high concentration of chloride could compromise the chromatography.This method can be used for Cr(Ⅵ) determination in groundwater.
ion chromatography;post-column derivatization;groundwater;Cr(Ⅵ)
徐硕,女,1990年出生,工程师,硕士研究生,从事环境监测工作,E-mail:shuoxu12@163.com。
刘保献,男,1983年出生,高级工程师,硕士研究生,从事环境监测工作,E-mail:liubaoxian28@163.com。
10.3936/j.issn.1001-232x.2016.06.008
2016-03-14
