阴离子、阳离子和非离子表面活性剂在纳/微米草酸钙晶体上的吸附差异
2016-12-20温小玲丁一鸣欧阳健明
温小玲 丁一鸣 欧阳健明
阴离子、阳离子和非离子表面活性剂在纳/微米草酸钙晶体上的吸附差异
温小玲丁一鸣欧阳健明*
(暨南大学生物矿化与结石病防治研究所;暨南大学化学系,广州510632)
研究了尺寸分别为50 nm和3μm的一水草酸钙(COM)和二水草酸钙(COD)晶体对不同电荷表面活性剂的吸附差异,包括阴离子型表面活性剂磺基琥珀酸钠二辛酯(AOT)、阳离子型表面活性剂十六烷基三甲基溴化铵(CTAB)和非离子型表面活性剂壬基酚聚氧乙烯醚(NP-40),探究了尿液中带不同电荷的分子对纳米、微米尿微晶的影响。结果表明,表面活性剂的吸附量大小为AOT>CTAB>NP-40,即阴离子型表面活性剂的吸附量最大,非离子型表面活性剂的吸附量最小;晶体尺寸相同时,COM的吸附能力稍大于COD;吸附表面活性剂后,晶体表面的ζ电位绝对值都增大,有利于抑制晶体的团聚和沉降。提出了晶体吸附不同表面活性剂的分子模型。不同电荷表面活性剂与纳/微米COM、COD晶体之间存在不同的相互作用。表面活性剂吸附量越大,沉降越慢,对晶体悬浮液的稳定效果越明显。
晶体尺寸;阴离子表面活性剂;阳离子表面活性剂;非离子表面活性剂;吸附模型;草酸钙+1
A
1001-4861(2016)12-2102-07
O614.23
0 引言
草酸钙有3种水合物形式,即一水草酸钙(COM)、二水草酸钙(COD)和三水草酸钙(COT),其中COM和COD是尿结石的主要成分[1];COT由于容易在溶液中发生晶相转变,在结石中很少发现。作为生物体内常见的无机矿化产物之一,草酸钙晶体还广泛存在于高等植物体内,参与植物细胞内钙离子的调控[2]。草酸钙还有利于形成致密生物矿化膜,达到保护石质文物的目的。
在人体尿液中存在大量的有机物质,包括氨基酸、柠檬酸、胆汁盐、白蛋白、酸性粘多糖及磷脂等。其中的不少阴离子物质具有表面活性;而当尿液pH值降低时,会引起部分蛋白质和氨基酸带正电荷,使得其具有类似阳离子表面活性剂的性质。尿液中的表面活性物质能降低尿液的表面张力,通过钙桥和其他作用吸附到草酸钙上,影响尿液中草酸钙的成核、生长、聚集以及晶相的转变,最终影响结石的形成[3-6]。
表面活性剂具有自组装和易于吸附到界面的性质,是系统研究有机物分子调控晶体形成和转化的理想模型分子[5]。磺基琥珀酸钠二辛酯(AOT)、十六烷基三甲基溴化铵(CTAB)和壬基酚聚氧乙烯醚(NP-40)分别为阴离子型、阳离子型和非离子型表面活性剂,它们的化学性质稳定,在与晶体的吸附过程中没有H+离子释放,因此,可以忽略体系pH值改变带来的干扰,得到的结果主要反映表面活性剂本身性质的影响;并且,AOT、CTAB和NP-40是常用的研究表面活性剂对生物矿物(如草酸钙、磷酸钙和磷酸氢钙)影响的代表性物质[7-9]。例如,AOT可以优先吸附在COM的(101)晶面上,因此,AOT不但能抑制或延缓COM晶体的生长,还会促进COD晶体的形成[9];CTAB可诱导蝴蝶状COM晶体形成,且随着CTAB浓度增加,晶体尺寸减小[7]。非离子型表面活性剂(C16EO8)能聚集草酸根离子,从而促进COM的成核,减缓草酸钙晶体的长大[9]。
草酸钙结石的形成包括晶体的成核、生长、聚集及晶体在体内的滞留等过程。之前的报道注重研究各种添加剂对草酸钙晶体生长和晶相转变的影响,而对尿液中的表面活性物质(如蛋白质、胆汁盐)在不同尺寸、不同晶相的草酸钙晶体表面的吸附及其影响研究较少。尿液中同时存在纳米和微米级晶体[10-12],且存在不同性质的表面活性物质。尿微晶的尺寸不同,其比表面积、表面能和表面所带的电荷量等都存在差异,从而影响其与表面活性物质的吸附量[13],最终影响结石的形成。例如,带正电荷的纳米铈能吸附更多带负电荷的牛血清白蛋白(BSA),而带负电荷的纳米铈吸附BSA较少[14]。但至今为止,还没有同时比较阴离子、阳离子和非离子表面活性剂在纳米/微米晶体表面吸附差异的报道。
我们已报道了尺寸从50 nm到20μm的系列COM和COD晶体[15],并研究了它们的理化性质;对NP-40的吸附研究表明,尺寸小于1μm(包括50、100 nm、1μm等)晶体的吸附曲线均相近(为直线型),而尺寸3、10和20μm晶体的吸附曲线也相近(均为S型)[16]。基于前期对尿微晶[11-12]和NP-40的研究结果,本文选择50 nm和3μm两个尺寸的晶体作为代表性的纳米和微米级尿微晶,选取AOT、CTAB和NP-40分别代表尿液中带负电荷、带正电荷和不带电荷的表面活性组分,比较研究这些晶体与不同表面活性剂的吸附特性及其差异,以期进一步研究不同荷电分子对生物矿物形成的影响,并探讨尿液中不同表面活性物质影响草酸钙结石形成的机理。
1 实验材料与方法
1.1试剂与仪器
磺基琥珀酸钠二辛酯(AOT,上海楷洋生物技术有限公司)、十六烷基三甲基溴化铵(CTAB,天津市大茂化学试剂公司)、壬基酚聚氧乙烯醚(NP-40,上海阿拉丁试剂有限公司)、溴百里酚蓝、亚甲基蓝、碘化钾、碘和其他常规试剂均为分析纯试剂。实验用水均为二次蒸馏水。
尺寸50 nm和3μm的COM、COD晶体参照文献[15]合成,X射线粉末衍射(XRD)和红外光谱(FTIR)表明合成的晶体为纯的目标产物。
Zetasizer Nano-ZS型纳米粒度-Zeta电位分析仪(英国Malvern公司)。D/max2400X射线粉末衍射仪(日本理学)。傅里叶变换红外光谱仪(美国Nicolet公司)。500型紫外可见分光光度计(美国Varian Cary公司)。
1.2吸附实验
在一系列的25 mL烧杯中加入COM或COD晶体50 mg,分别加入预先配置好的浓度(c0)分别为0、30、60、90、120、150、200、250、300、350、450 mg·L-1的不同表面活性剂(AOT、CTAB或NP-40)标准溶液15 mL,此时溶液pH值为6.6±0.1;超声10 min至晶体完全分散,密封后放入37℃恒温箱中吸附24 h,离心后,取上清液测定剩余表面活性剂浓度ceq,同时测量悬浮液的ζ电位。根据公式(1)计算晶体对表面活性剂的吸附量Qads。以c0为横坐标,Qads为纵坐标绘制吸附曲线。AOT检测采用亚甲基蓝方法[17],其线性方程为:y=0.1137x-0.0149,R2=0.9985;CTAB采用溴百里酚蓝方法测定[18],其线性方程为:y= 0.0542x-0.0404,R2=0.9997;NP-40采用碘化钾-碘方法测定[19],其线性方程为y=0.0072x-0.0104,R2= 0.9985。

选择表面活性剂的浓度为0~450 mg·L-1是基于AOT、CTAB和NP-40在水溶液中的临界胶束浓度(CMC)分别为6.8×10-4、6×10-4和5.09×10-4mol· L-1[20-22],即分别为302.3、218.6和367.0 mg·L-1,实验浓度涵盖了3种表面活性剂的CMC。
1.3草酸钙晶体的稳定性实验
称取5 mg的COM或COD晶体于4个25 mL的烧杯中,分别加入蒸馏水或浓度为150 mg·L-1的AOT、CTAB和NP-40溶液,超声5 min至完全分散,用UV-Vis分光光度计在波长620 nm处[23]测定COM、COD悬浊液在静置不同时间后的光密度(At),并参照文献[24]中聚集系数的计算方法,将静置t min (t=30或5 min)的光密度(At值)与静置前(t=0 min)的光密度(A0)的差值称为沉降系数(SC),即:
SC=(A0-At)×100(2)
式中100是为了使SC由小数转化为整数,SC越大表示悬浊液中晶体的沉降程度越大。
2 结果与讨论
2.1微米和纳米草酸钙的吸附曲线
图1为不同表面活性剂在尺寸分别为50 nm和3μm的COM、COD晶体表面的吸附曲线,图2为在表面活性剂浓度c=450 mg·L-1时,纳/微米COM、COD晶体对不同表面活性剂的最大吸附量。从图1和图2可以看出:
(1)无论是COM还是COD晶体,吸附量的大小顺序均为:AOT>CTAB>NP-40。即阴离子表面活性剂的吸附量最大,非离子表面活性剂吸附量最小,阳离子表面活性剂处在两者之间。
AOT含酸性基团,分子带负电荷,其与草酸钙晶体存在3种作用:一是AOT与带正电荷的COM之间存在较强的静电吸引力;二是AOT能与晶体表面暴露的钙离子(即正电荷位点)直接连接[25],形成(Ca(Ox))Ca2+…-O3S(AOT)键(图3中);三是AOT可模拟草酸根的取向与草酸钙晶体形成钙桥[26],接到晶体表面的负电荷位点上,即通过钙离子形成(Ca(Ox))COO-…Ca2+…-O3S(AOT)键。由于AOT在纳/微米COM、COD晶体上的吸附位点较多,静电吸引力大,因此AOT的吸附能力最强,吸附量也最大。
CTAB分子带正电荷,其阳离子基团可直接与草酸钙晶体表面的草酸根(即负电荷位点)结合[25-26],形成(Ca(Ox))COO-…+(H3C)3N(CTAB)键(图3左)。
非离子表面活性剂NP-40分子不带电荷,只能通过氢键吸附到COM、COD晶体表面(图3右),即形成(Ca(Ox))COO-…HO-(NP-40)键[16]。由于氢键的作用力比静电吸引力弱,因此NP-40的吸附能力弱于离子型表面活性剂AOT和CTAB[27],吸附量最小。
(2)对于相同尺寸的晶体,COM晶体的吸附能力均稍大于COD。这是因为COM晶体表面同时具有带正电荷和负电荷的区域,COM晶体(101)面暴露出大量的钙离子,而(010)面上的草酸根离子相对较多。COD的形貌是四角双锥型,有8个相等的(101)和4个相等的(100)面,(101)和(100)面都包含着高浓度的水分子,这减少了晶面的电荷;COD唯一的高电荷区是双锥体的2个顶端,但这只占总表面积的一小部分[9],因此,COD表面用于吸附的位点会比COM的少。
(3)同种晶体,纳米级晶体对表面活性剂的吸附量远大于微米晶体。这是因为纳米颗粒的比表面积大,表面原子数目和可吸附表面活性剂的位点均急剧增加,因此,吸附量显著增大[28];
虽然AOT、CTAB和NP-40的尾链长度不同,但文献[29-30]报道尾链对吸附的影响较小。例如,Rosen[29]比较研究了烷基苯磺酸钠和烷基磺酸钠在Al2O3等固体表面的吸附,表明疏水链的长短和数量对吸附量影响不大。吸附量主要由亲水基决定;亲水基团越大,表面饱和吸附量越小;而亲水基数量增加后,由于伴随着亲水基的横截面积增大,每个分子占据的固体表面积增大,也会导致吸附量减小[30]。
(4)纳米COM、COD晶体的吸附曲线呈直线型(图1A),而微米COM、COD晶体的吸附曲线呈S型(图1B)。这是因为纳米晶体的比表面积远大于微米晶体,即使在实验的最大表面活性剂浓度(c=450mg·L-1)时,纳米晶体仍然没有达到吸附平衡状态(图1A),即这些纳米晶体表面仍然有未被表面活性剂占据的位点。相比之下,微米晶体由于形成双分子层或胶束等原因[16],在c=200~300 mg·L-1时,3μm COM和COD的吸附就已先后达到吸附平衡(图1B),导致S型吸附曲线的形成。
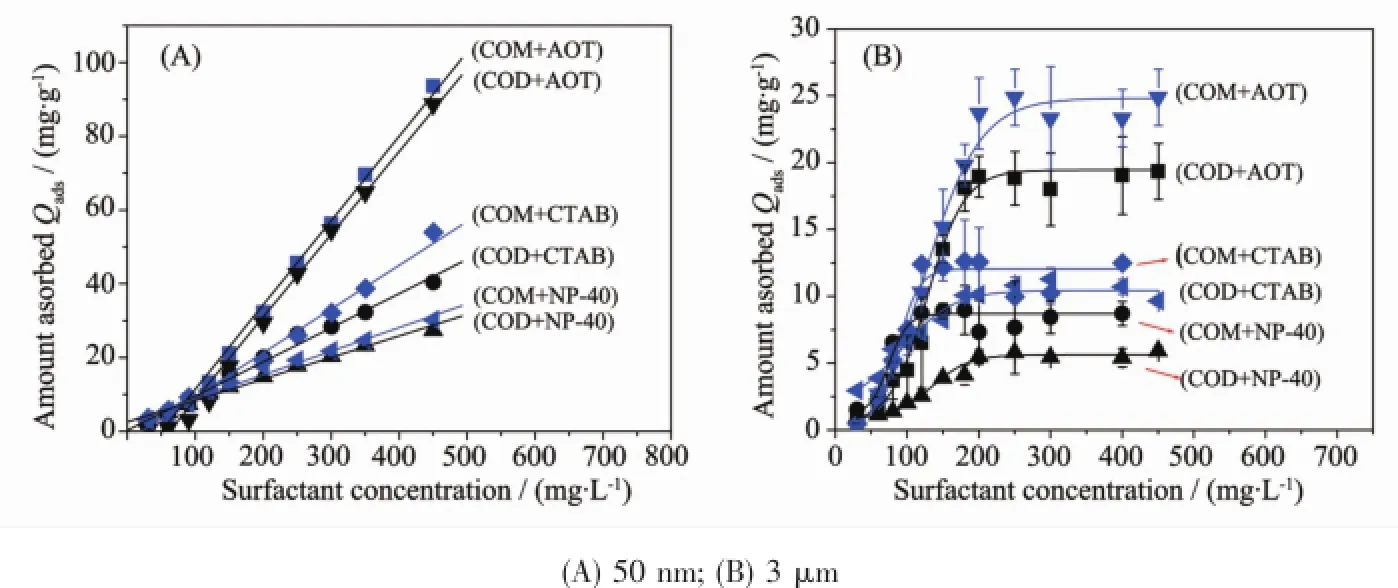
图1 不同表面活性剂在纳/微米COM、COD晶体上的吸附曲线Fig.1 Adsorption curves of different types of surfactants on micron/nano COM and COD
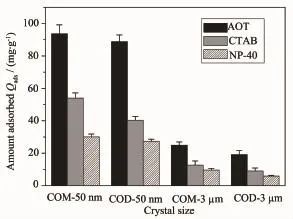
图2 纳/微米草酸钙晶体对不同表面活性剂的最大吸附量Fig.2 Maximum adsorption quantity of different types of surfactants on micron/nano calcium oxalate crystals
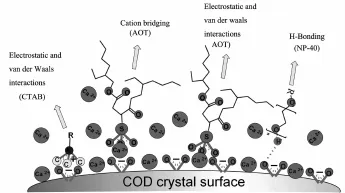
图3 COD晶体吸附不同表面活性剂的分子模型(COM晶体与COD的相近)Fig.3 Schema of different types of surfactants adsorbed on COD crystal(COM crystal is similar to COD)
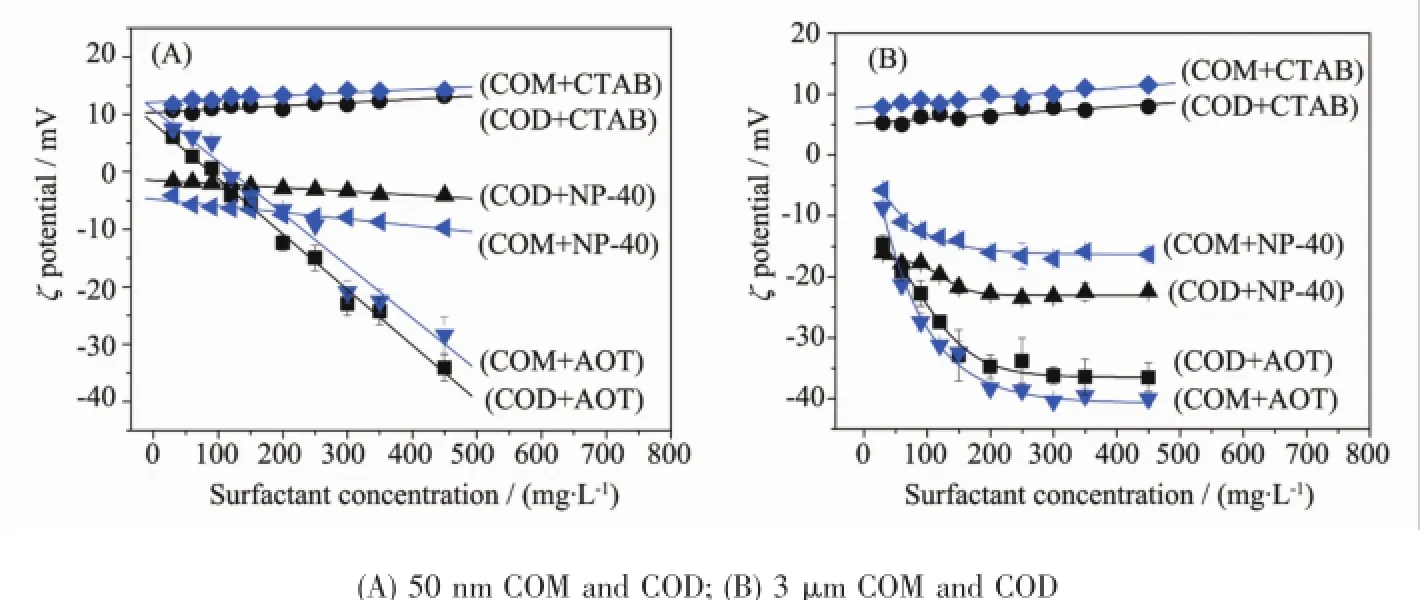
图4 纳/微米COM、COD晶体ζ电位随不同表面活性剂浓度的变化Fig.4ζpotential change of nano/micron COM and COD crystals with concentrations of different types of surfactants
2.2COM和COD晶体吸附不同表面活性剂后的ζ电位变化
图4为纳/微米COM、COD吸附不同表面活性剂后的ζ电位变化,其变化趋势与表面活性剂的性质密切相关。AOT分子带负电荷,所以晶体吸附AOT后,其表面的负电荷增加,ζ电位变负,且由于AOT的吸附量最大,因此,在最大吸附量时AOT引起的晶体ζ电位绝对值增加幅度也最大。
而CTAB分子带正电荷,因此,晶体吸附CTAB后其表面的正电荷增加,ζ电位变正;但由于CTAB在COM和COD晶体表面的最大吸附量均明显小于AOT(图2),仅为AOT吸附量的一半左右,因此,CTAB引起的晶体ζ电位变化幅度小于AOT。
NP-40为不带电荷的中性分子,因此,吸附NP-40后晶体的ζ电位变化最小。但由于NP-40在通过氢键吸附到晶体表面时,晶体表面的电荷密度也发生了改变,导致吸附NP-40后晶体的ζ电位绝对值稍有降低。
ζ电位的变化差异进一步验证了纳/微米COM、COD对不同表面活性剂的吸附。
2.3吸附不同表面活性剂后COM和COD晶体的沉降
通过检测溶液光密度值随时间的变化,研究了纳/微米COM、COD晶体分散在不同表面活性剂中的沉降。加入表面活性剂后,沉降系数(SC)均变小(图5),且晶体的沉降速度与表面活性剂的吸附量呈负相关。说明表面活性剂吸附在草酸钙晶体上,使草酸钙悬浮液的稳定性增加。加入不同表面活性剂后晶体沉降速度由慢到快依次为AOT、CTAB和NP-40,即:表面活性剂吸附量越大,对晶体悬浮液的稳定效果越好。
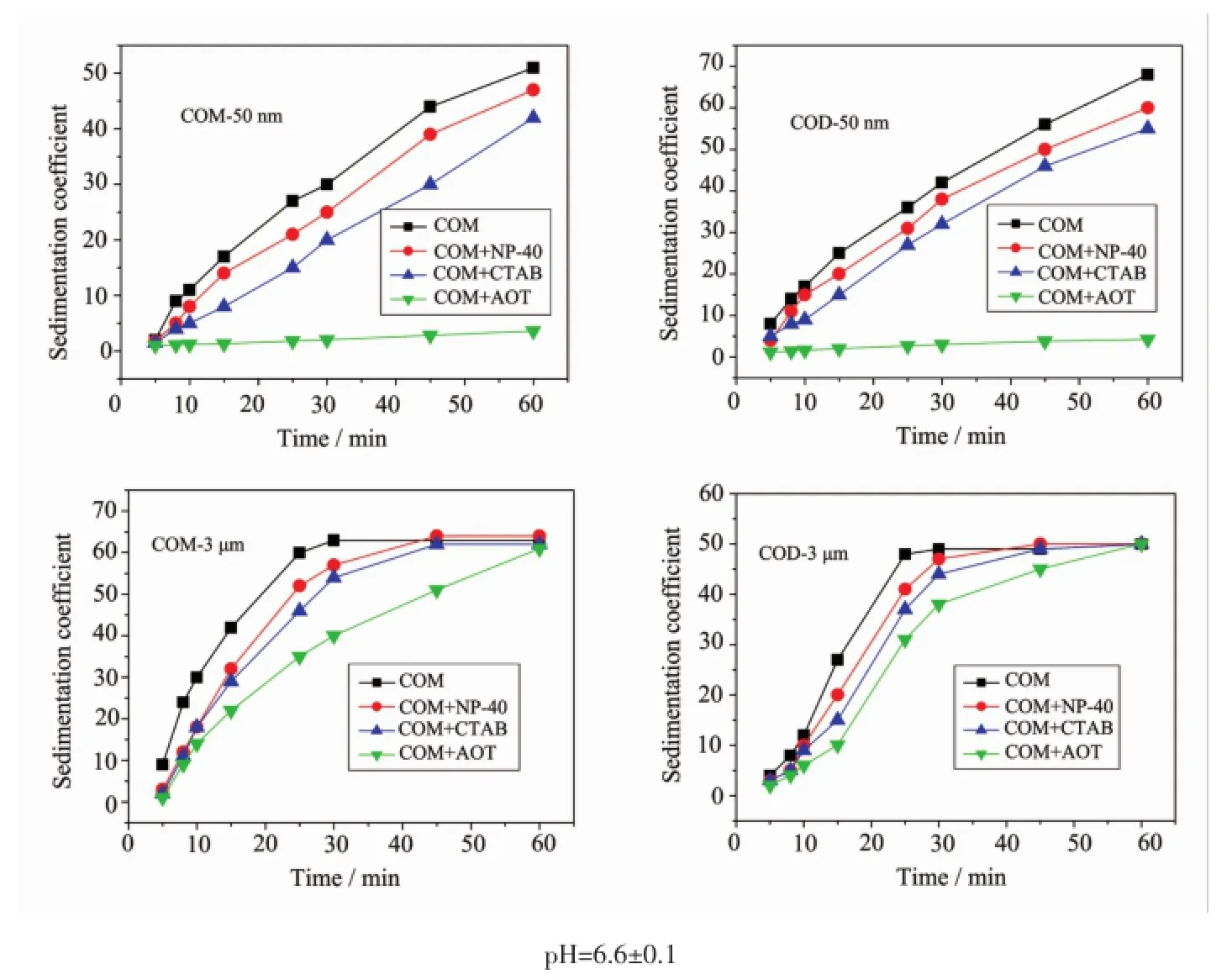
图5 纳/微米COM、COD沉降系数随不同表面活性剂的变化Fig.5 Change of sedimentation coefficient of micron/nano COM and COD crystals different with surfactants
晶体的沉降速度还与晶体的ζ电位绝对值呈负相关(表1)。尺寸相同时,晶体ζ电位绝对值越大,沉降越慢,其对应的SC越小。例如,50 nm COM晶体的ζ电位绝对值(2.20 mV)大于COD(1.5 mV),故其在加入表面活性剂前后的SC值均小于COD;而微米晶体会比纳米晶体沉降快还归因于微米晶体的重力作用比纳米晶体的大。
根据吸附曲线(图1)、ζ电位(图4)和沉降系数变化(图5),我们提出了纳/微米草酸钙晶体的吸附机理如下:
对于微米晶体,其吸附AOT和CTAB后的静电排斥作用模型如图6所示。微米晶体的吸附曲线呈S型(图1B)。S型吸附曲线分可为3个阶段[16,27]:在阶段1,即cAOT<100 mg·L-1或cCTAB<60 mg·L-1时,微米晶体的吸附量随cAOT或cCTAB增加而缓慢增加,此时AOT或CTAB在固体表面发生单分子层吸附(图6a)。在阶段2,即100<cAOT<200 mg·L-1或60<cCTAB<150 mg·L-1时,吸附曲线斜率增大,此时形成大量的不完整的双分子层或扁平的胶束,导致吸附量迅速增加,造成此阶段吸附曲线斜率较高。在阶段3:即cAOT>200 mg·L-1或cCTAB>150 mg·L-1时,吸附量的增加速度再次变缓。因为形成了紧密的双分子层或胶束,表面活性剂带电荷的亲水基一端暴露在水中(图6b),使得溶液中的带电荷的表面活性剂分子与晶体表面的库伦斥力越来越大,吸附量增加变缓,并在cAOT=220 mg·L-1或cCTAB=180 mg·L-1左右时,吸附逐渐达到饱和,导致S型吸附曲线的形成。并且,形成的这种带电荷的、被表面活性剂包裹的晶体会因为静电斥力而难以团聚和沉降。
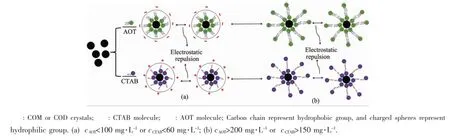
图6 微米草酸钙晶体吸附AOT或CTAB后的静电排斥作用Fig.6 Electrostatic repulsion of micron Ca(Ox)crystals after adsorption of AOT or CTAB

表1 纳/微米COM、COD在水中的Zeta电位及添加不同表面活性剂后的沉降系数Table 1 Zeta potential of micron/nano COM and COD crystals in aqueous solution and sedimentation coefficient(SC) after addition of various surfactants
对于纳米晶体,由于在实验所用表面活性剂的浓度范围内(0~450 mg·L-1)未达到饱和,纳米COM、COD晶体的吸附曲线呈直线型(图1A)。但吸附表面活性剂后也会增大晶体的ζ电位绝对值(图4),使得晶体之间存在的静电斥力作用增大,阻止纳米COM和COD晶体的团聚,这从图5中加入表面活性剂后沉降系数减小亦可得到证实。
前文[31]我们测量了纳米/微米COM和COD晶体ζ电位随pH值的变化。随着体系pH值增大,晶体表面所带负电荷逐渐增大,使得晶体的ζ电位由正逐渐变负,且纳米晶体的等电点(晶体所带净电荷为零时的pH值)大于微米晶体的等电点。当pH值小于晶体等电点时,晶体ζ电位随pH值增大而减小,此时pH值增大会减小晶体之间的静电斥力,增加草酸钙晶体的聚沉[32];而pH值大于晶体等电点时,晶体ζ电位随pH值增大而不断变负,即ζ电位的绝对值不断增大,此时pH值增大会增大晶体之间的静电斥力,因而减小草酸钙微晶聚沉。在pH值处于晶体等电点时,晶体表面不带电荷,晶体之间不存在静电斥力作用,此时草酸钙晶体聚沉最严重。
图5为在pH=6.6±0.1时的研究结果,此pH值低于纳米晶体的等电点,但高于微米级晶体的等电点。此时如果pH值出现少许降低,由于纳米级晶体ζ电位会增加,晶体间的静电斥力增加,会减轻晶体的聚沉;而微米晶体则由于ζ电位绝对值降低,晶体间的静电斥力减小,会加重微晶的聚沉。
由于本文采用的3种表面活性剂(AOT、CTAB和NP-40)在与草酸钙晶体吸附后均没有H+离子的释放,因此,不同浓度的表面活性剂加入后,只是影响了溶液的离子强度,溶液的pH值在吸附前后变化不大,故pH值对吸附的影响可以忽略不计。
由于肾小管中小尺寸晶体在滞留期间(约10 min)不足以生长到足够大堵塞肾小管,因此晶体的聚集才是导致尿微晶尺寸迅速增大的重要原因[33-34]。本文结果表明,吸附表面活性剂后可降低晶体的聚集,增加晶体悬浮液的稳定性,推测通过添加带负电荷的表面活性物质来改变尿微晶的荷电性质,有可能达到抑制尿结石的形成。
3 结论
纳/微米COM、COD晶体对不同表面活性剂的吸附量大小顺序为AOT>CTAB>NP-40。不同表面活性剂与纳/微米COM、COD晶体之间的相互作用不同,导致吸附量不同。吸附表面活性剂后,晶体的ζ电位绝对值都增大,有利于抑制晶体的团聚;吸附表面活性剂后,沉降速度均减慢,沉降速度与晶体的ζ电位的绝对值和表面活性剂的吸附量呈负相关。
[1]Zhang D,Qi L,Ma J,et al.Chem.Mater.,2002,14(6):2450-2457
[2]Zhang G N,Ouyang J M,Xue J F,et al.Mater.Sci.Eng.C, 2013,33(7):4039-4045
[3]Sürer H,Ozgün T,Yilmaz F M,et al.Clin.Chem.Lab.Med., 2014,52(4):e71-e73
[4]Poon N W,Gohel M D I.Carbohyd.Res.,2012,347:64-68
[5]Sikiric M,Filipovi-Vincekovic N,Babic-Ivancic V,et al.J. Colloid Interface Sci.,1999,212(2):384-389
[6]Saso L,Grippa E,Gatto M T,et al.Int.J.Urol.,2001,8(3): 124-127
[7]Wei X X,Yang J,Li Z Y,et al.Colloids Surf.A,2012,401: 107-115
[8]Dong R,Weng R,Dou Y,et al.J.Phys.Chem.B,2010,114 (6):2131-2139
[9]Sikiric M D,Füredi-Milhofer H.Adv.Colloid Interface Sci., 2006,128:135-158
[10]Elliot J S,Rabinowitz I N.J.Urol.,1980,123(3):324-327
[11]Gao J,Xue J F,Xu M,et al.Int.J.Nanomed.,2014,9:4399-4409
[12]LIU Yi-Ming(柳一鸣),XIA Zhi-Yue(夏志月),OUYANG Jing-Ming(欧阳健明),et al.Chinese J.Inorg.Chem.(无机化学学报),2013,29(5):903-909
[13]Suttiponparnit K,Jiang J,Sahu M,et al.Nanoscale Res. Lett.,2011,6,1-8
[14]Patila S,Sandbergb A,Heckertc E,et al.Biomaterials, 2007,28(31):4600-4607
[15]Sun X Y,Ouyang J M,Liu A J,et al.Mater.Sci.Eng.C, 2015,57:147-156
[16]WEN Xiao-Ling(温小玲),GAN Qiong-Zhi(甘琼枝), OUYANG Jing-Ming(欧阳健明).Chinese J.Inorg.Chem. (无机化学学报),2015,31(10):2021-2029
[17]Tunik L,Furedi-Milhofer H,Garti N.Langmuir,1998,14: 3351-3355
[18]Chebotarev A N,Paladenko T V,Shcherbakova T M.J. Anal.Chem.,2004,59(4):309-313
[19]Brown D G,Jaffe P R.Environ.Sci.Technol.,2001,35: 2022-2025
[20]Tunik L,Addadi L,Garti N,et al.J.Cryst.Growth,1996, 167(3):748-755
[21]Mandal A B,Nair B U.J.Phys.Chem.,1991,95(22):9008-9013
[22]Calvo E,Bravo R,Amigo A,et al.Fluid Phase Equilib., 2009,282(1):14-19
[23]Kulaksizoglu S,Sofikerim M,Cevik C.Int.J.Urol.,2007,14 (3):214-218
[24]Christmas K G,Gower L B,Khan S R,et al.J.Colloid Interface Sci.,2002,256(1):168-174
[25]Sheng X,Jung T,Wesson J A,et al.Proc.Natl.Acad.Sci. U.S.A.,2005,102(2):267-272
[26]Farmanesh S,Chung J,Sosa R D.J.Am.Chem.Soc.,2014, 136(36):12648-12657
[27]Zhang R,Somasundaran P.Adv.Colloid Interface Sci.,2006, 123:213-229
[28]Wang X,Jiang Z Y,Xie Z X,et al.Acc.Chem.Res.,2014, 42(7):308-318
[29]Rosen M J.J.Am.Oil Chem.Soc.,1975,52(11):431-435
[30]Paria S,Yuet P K.Ind.Eng.Chem.Res.,2007,46(1):108-113
[31]WEN Xiao-Ling(温小玲),OUYANG Jing-Ming(欧阳健明). Chinese J.Inorg.Chem.(无机化学学报),2016,32(1):103-110
[32]Ofir E,Oren Y,Adin A.Desalination,2007,204(1):33-38
[33]Finlayson B,Reid S.Invest.Urol.,1978,15:442448
[34]Kok D J,Khan S R.Kidney Int.,1994,46:847
Differences in Adsorption of Anionic,Cationic and Nonionic Surfactant on Micron/nano Calcium Oxalate Monohydrate and Dihydrate Crystals
WEN Xiao-Ling DING Yi-Ming OUYANG Jian-Ming*
(Institute of Biomineralization and Lithiasis Research,Jinan University,Guangzhou 510632,China)
In order to explore the effect of urine components with different charges on micron/nano urinary crystallites,the adsorption difference of three surfactants with different charges onto micron/nano calcium oxalate monohydrate(COM)and dihydrate(COD)was studied,including anionic surfactant sodium diisooctyl sulfosuccinate(AOT),cationic surfactant cetyltrimethylammonium bromide(CTAB),and nonionic surfactant nonylphenol ethoxylate(NP-40).The adsorption quantity of surfactants on COM and COD was ranked in the following order:AOT>CTAB>NP-40,that is,the absorption quantity of anionic surfactant is the maximum,and absorption quantity of nonionic surfactantis the minimum.The adsorption quantity of COM was greater than COD with the same size;the absolute value ofζpotential on surface of COM and COD was increased after adsorption of surfactants,it was conducive to inhibit crystal aggregation and sedimentation.The molecular models of surfactants adsorbed on crystal surface were proposed.There are different interactions between micro/nano COM, COD crystal and anionic,cationic,nonionic surfactants.The larger the adsorption quantity of surfactants is,the slower the sedimentation rate is,so the stabilizing effecton crystalsuspension is more obvious.
crystal size;anionic surfactant;cationic surfactant;nonionic surfactant;adsorption model;calcium oxalate
10.11862/CJIC.2016.269
2016-04-16。收修改稿日期:2016-09-09。
国家自然科学基金(No.21371077)资助项目。
*通信联系人。E-mail:toyjm@jnu.edu.cn
